
丁寒锋教授团队JACS:计算化学修正结构辅助实现Elisapterane及生源相关二萜类天然产物的发散性全合成
近期,我系丁寒锋教授团队基于生源启发的末端D环构建思路,采用氧化去芳构化引发(ODI)的(5+2)环加成/1,2-酰基迁移策略,结合SmI₂介导的频哪醇偶联/Grob裂解/脱氧串联反应,高效构建了具有norneoelisabane型骨架的三环共同中间体。基于该策略,团队完成了包括elisapterosins A–F、aberrarone、elisabanolide及3-epi-elisabanolide等三大类九个天然产物的全合成。此外,研究还借助计算核磁技术,精准预见了elisapterane类天然产物家族成员间化学结构的罕见突变,成功完成了对elisapterosins A、D和F结构的修正,该结果最终通过全合成获得有力验证。相关研究成果发表于《Journal of the American Chemical Society》。
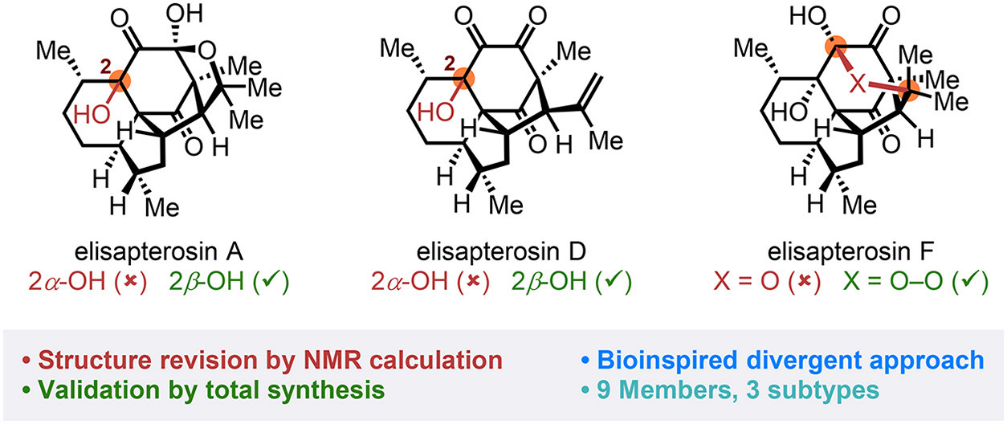
本文工作概述。图片来源:J. Am. Chem. Soc.
海洋是天然产物的丰富来源,蕴藏着众多结构复杂多样的化合物。这些结构新颖的海洋天然分子长期以来吸引着合成化学领域的广泛研究兴趣。然而,由于天然来源有限、提取困难,加之其本身结构复杂,其准确结构鉴定常常面临巨大挑战。文献中屡见不鲜的结构误判案例,也为有机化合物的结构解析工作提供了深刻教训。对从事天然产物全合成的化学家来说,投入大量精力合成一个最终被证明是错误的分子结构,无疑是令人十分挫败的经历。
历史上,曾多次出现通过“枚举式”全合成系统验证所有可能结构以修正天然产物真实构型的案例。客观而言,这类方法通常耗费巨大且成功率较低。如今,随着计算化学在有机化合物结构鉴定中的应用日益成熟,例如,借助计算化学方法可对有机分子的核磁共振谱这一指纹性信息实现可靠预测,该方法有望在合成前有效排除错误结构。与传统枚举合成策略相比,这种计算辅助手段在时间与资源成本上具有明显优势,并已在多个复杂天然产物的结构修订中取得良好效果(图一)。
此外,并非所有结构存疑的天然产物都适于采用枚举式全合成进行修正。汇聚式合成策略依赖于可独立制备并灵活组装的结构片段,为系统尝试多种结构变体提供了可能,但对于多环稠合类分子,实现有效的汇聚式切断往往较为困难。相应地,在线性合成路线中若依赖底物控制实现关键反应,一旦目标分子结构发生变动,很可能导致原有合成设计完全失效,需从头规划。
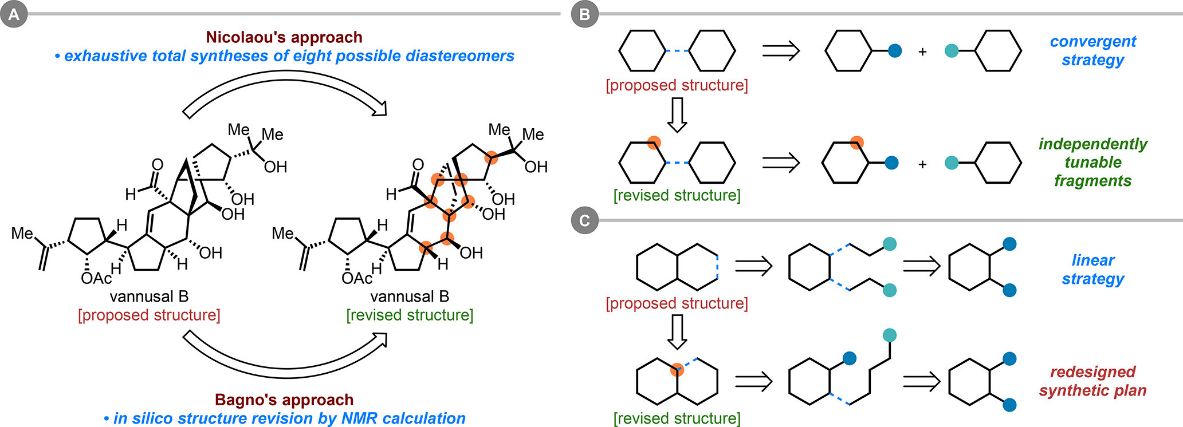
图一 图片来源:J. Am. Chem. Soc.
Elisapterane 二萜是由 Rodríguez 等人自Pseudopterogorgia elisabethae (Bayer)中分离得到的一类多环天然产物(图二)。目前,该家族已有六个成员陆续被分离和鉴定,分别为 elisapterosins A–F。初步研究发现,elisapterosin B 对结核分枝杆菌具有显著抑制作用。该标志性分子引起了多个国际合成团队的关注,其全合成研究也推动了一系列精巧策略与方法的发展。值得注意的是,以往在该类分子的合成中,对于环系的构建顺序存在相近的设计逻辑,即普遍倾向于优先构筑氧化程度较高的D环。本研究团队注意到elisapterane二萜在生源上与aberrarane及elisabane型二萜存在骨架连接方式的关联,进而提出了一种推迟构建D环的新策略,旨在通过基于neoelisabane型骨架的通用中间体,实现多个天然产物的发散式全合成。
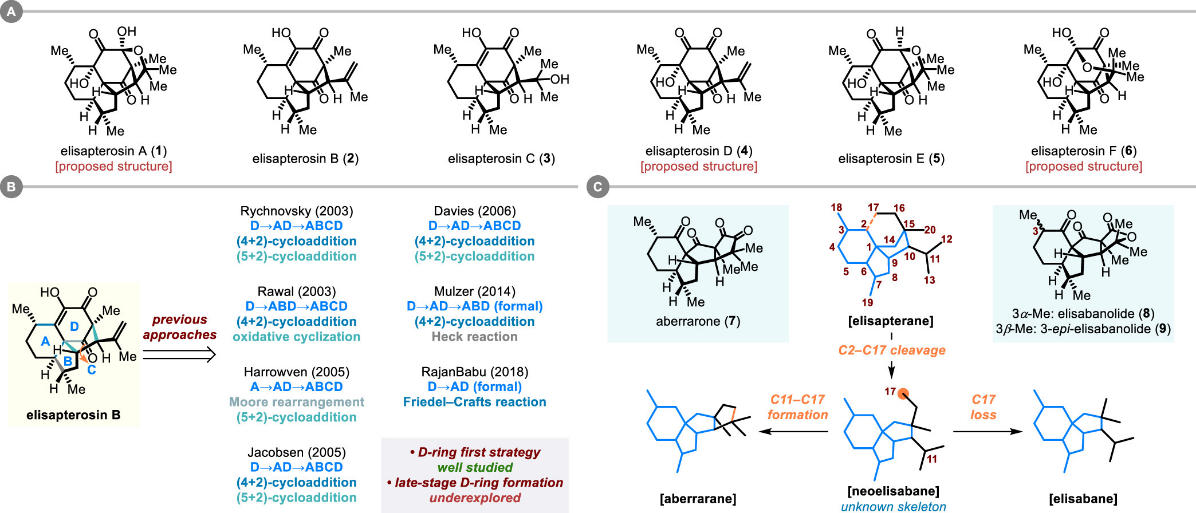
图二 图片来源:J. Am. Chem. Soc.
基于这一合成思路与课题组的前期工作,研究团队提出了基于ODI-(5+2) 环加成/1,2-酰基迁移及SmI2介导的频哪醇偶联/Grob裂解/去氧两组串联反应的逆合成分析(图三),靶向具有norneoelisabane型骨架的三环中间体(10)。
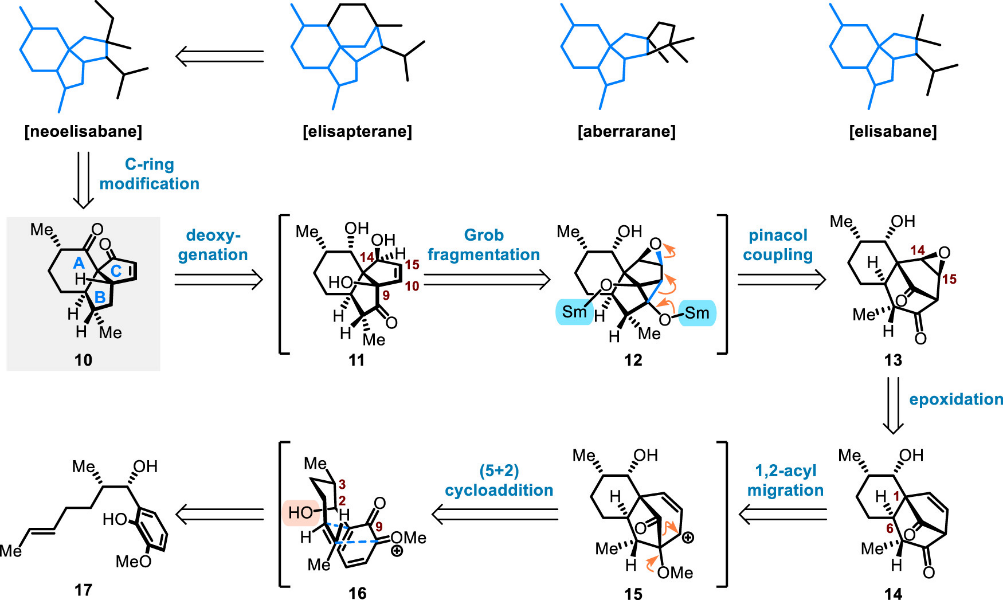
图三 图片来源:J. Am. Chem. Soc.
在对关键的SmI2介导的重排反应的探究中,研究团队基于对副产物生成的机理分析最终通过微调底物的方式成功克服了该串联反应的优化,随后顺利制备关键中间体(10)(图四、图五),在此基础上直接完成了aberrarone、elisabanolide及3-epi-elisabanolide的发散性合成(图六)。
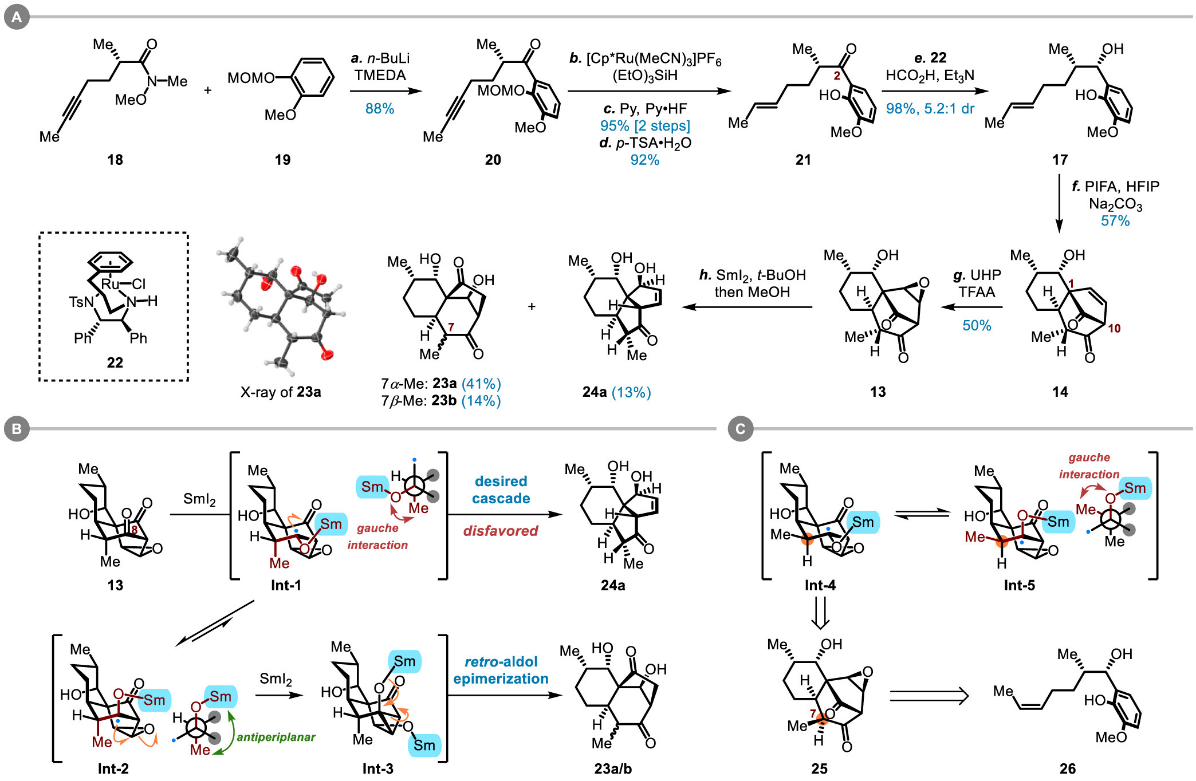
图四 图片来源:J. Am. Chem. Soc.
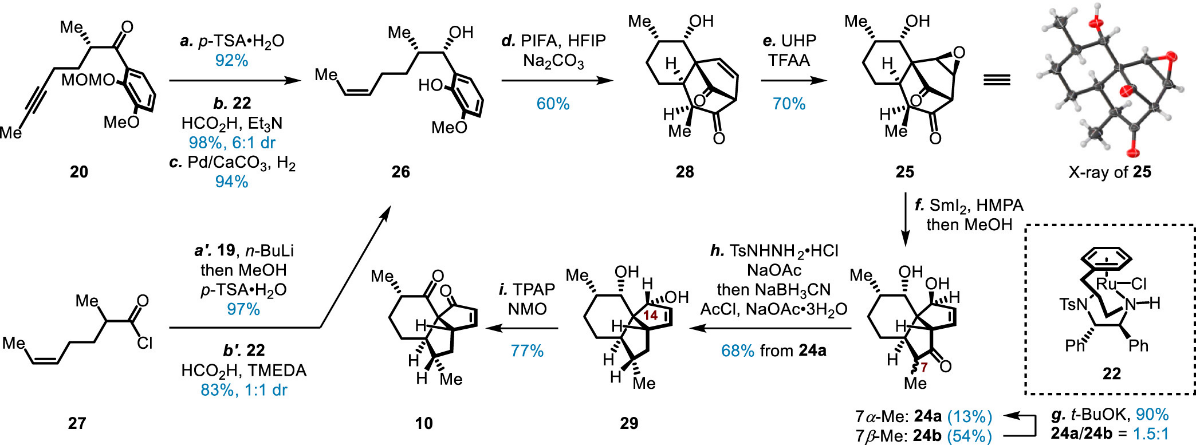
图五 图片来源:J. Am. Chem. Soc.
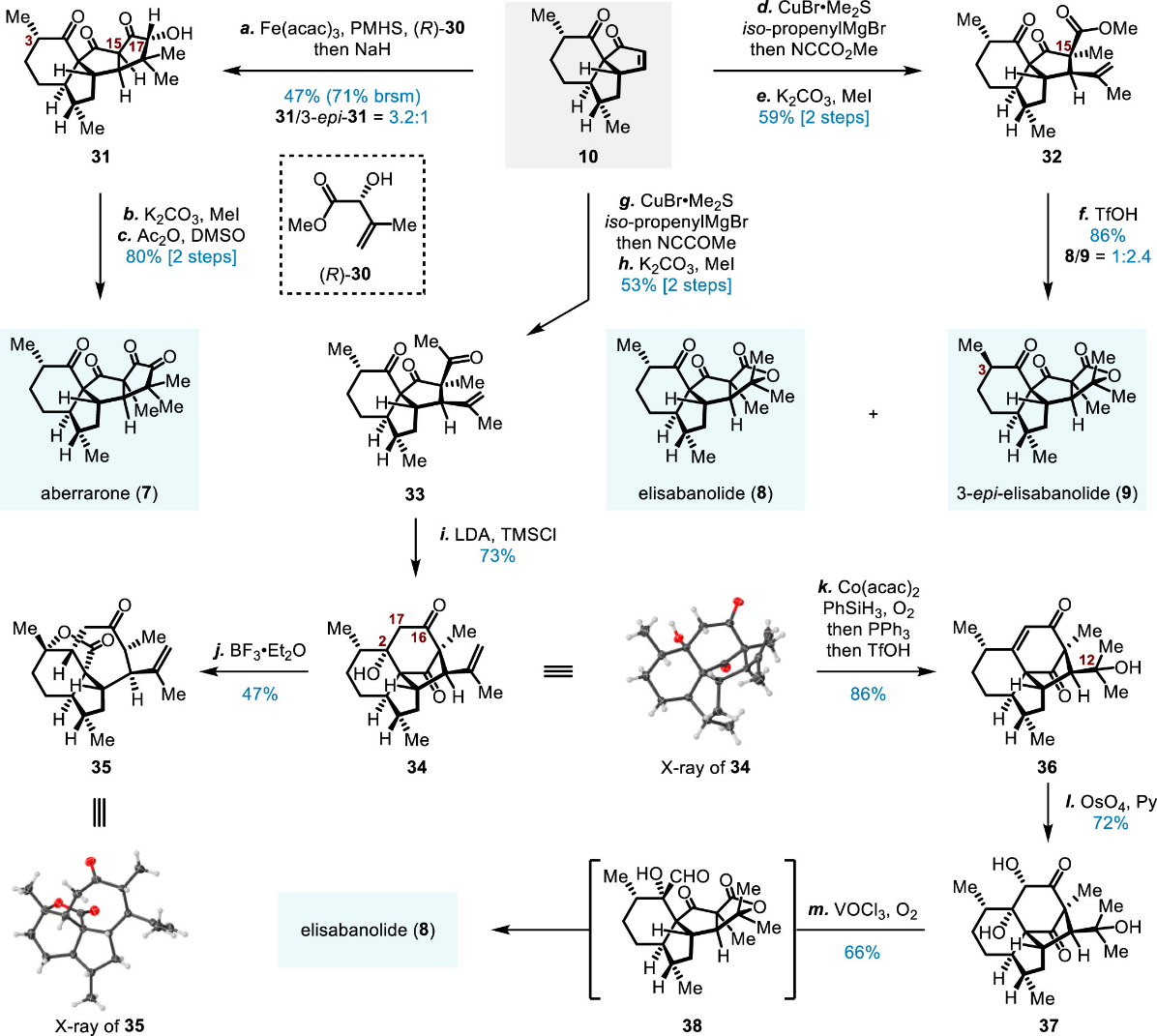
图六 图片来源:J. Am. Chem. Soc.
随后,课题组以四环中间体(37)为起点,进一步推进对elisapterane二萜的合成研究。期间,他们发现天然产物elisapterosin F的原有结构认定可能存在错误(图七)。通过重新审视多个迄今尚未完成合成的、结构更为复杂的elisapterane二萜(包括elisapterosins A、D、E和F)的原始测试数据,研究小组首先对分离文献中关于C2/C3位立体化学的指定提出了质疑。
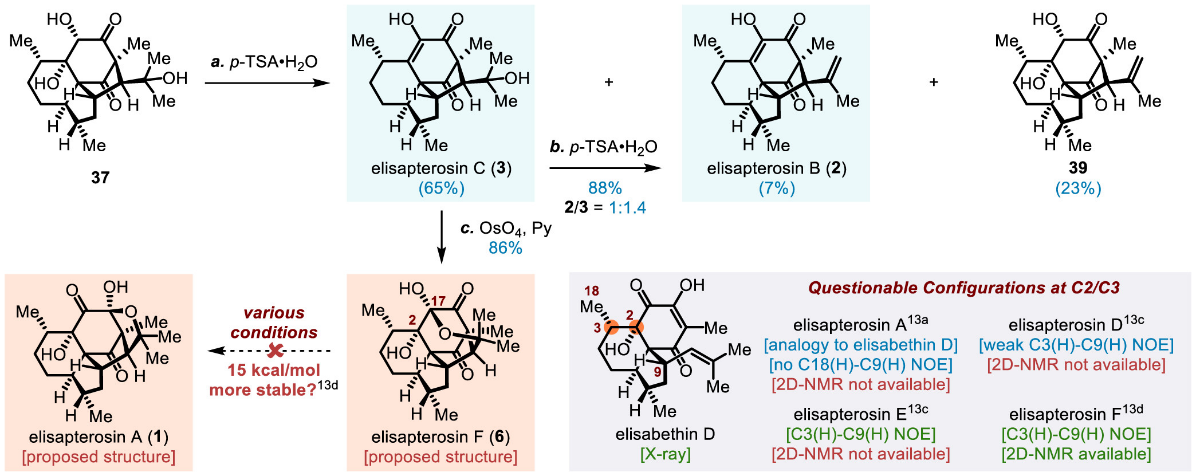
图七 图片来源:J. Am. Chem. Soc.
研究团队借助Sarotti小组发展的ANN-PRA与DP4+,以及中南大学王文宣团队建立的STS等核磁计算与分析手段,最终确定elisapterosins A和D中C2位羟基构型与分离文献中原先所提出的结构恰好相反(图八)。值得注意的是,在elisapterosin E中,原始文献所推断的构型正确无误,其C2位羟基的立体化学与推测结构极其相似的elisapterosin A截然相反。
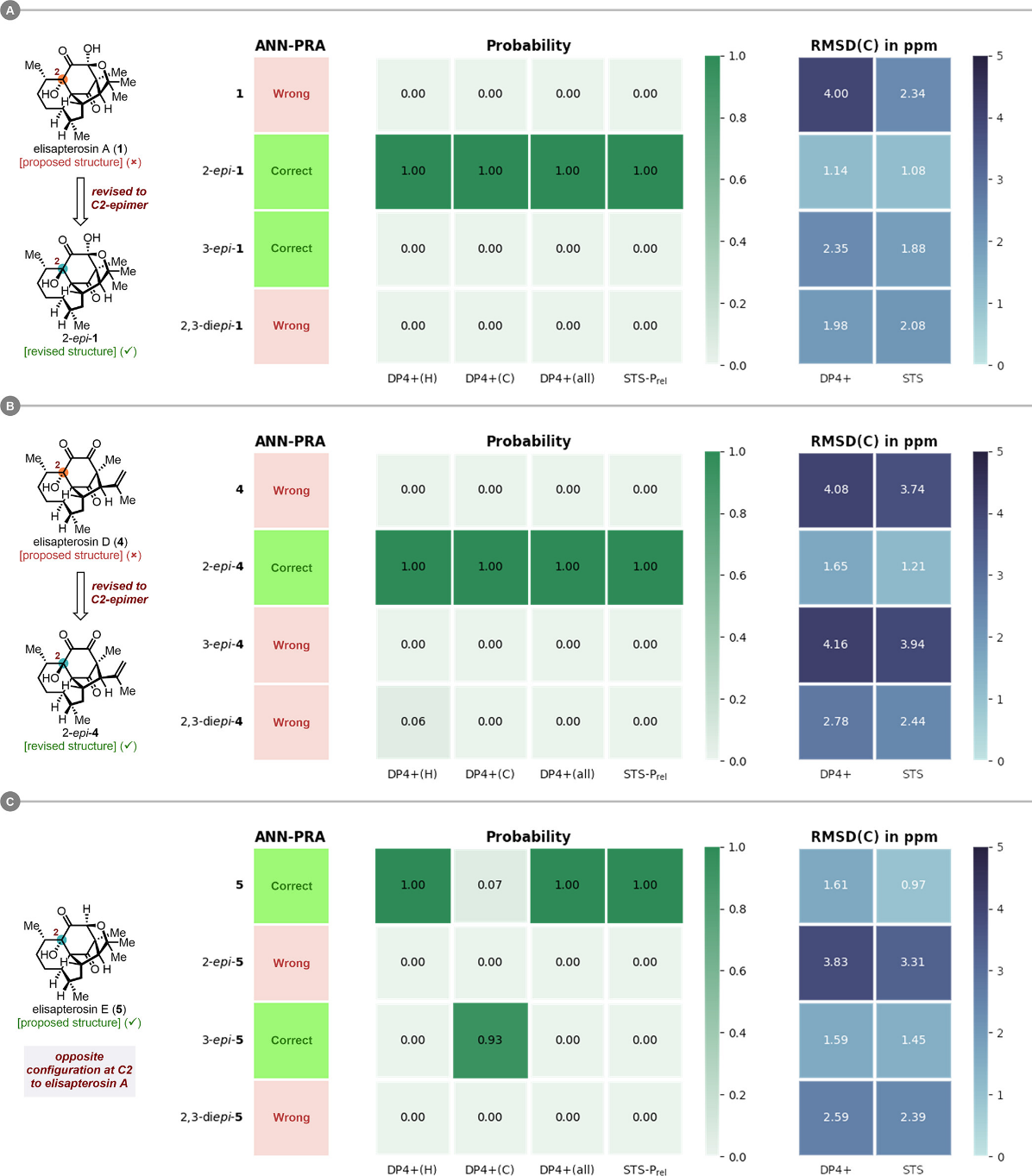
图八 图片来源:J. Am. Chem. Soc.
棘手之处在于,核磁计算显示elisapterosin F并非简单的C2/C3差向异构体(图九)。事实上,分离文献中提供的该分子二维图谱中,清晰的NOE信号关联也排除了这一可能性。研究团队一度被这个表面自洽实则矛盾的难题困扰,最终通过细致比对天然与合成样品之间碳谱数据的差异,并借助计算化学方法进行系统验证,提出其真实结构应为过氧半缩酮(41)。
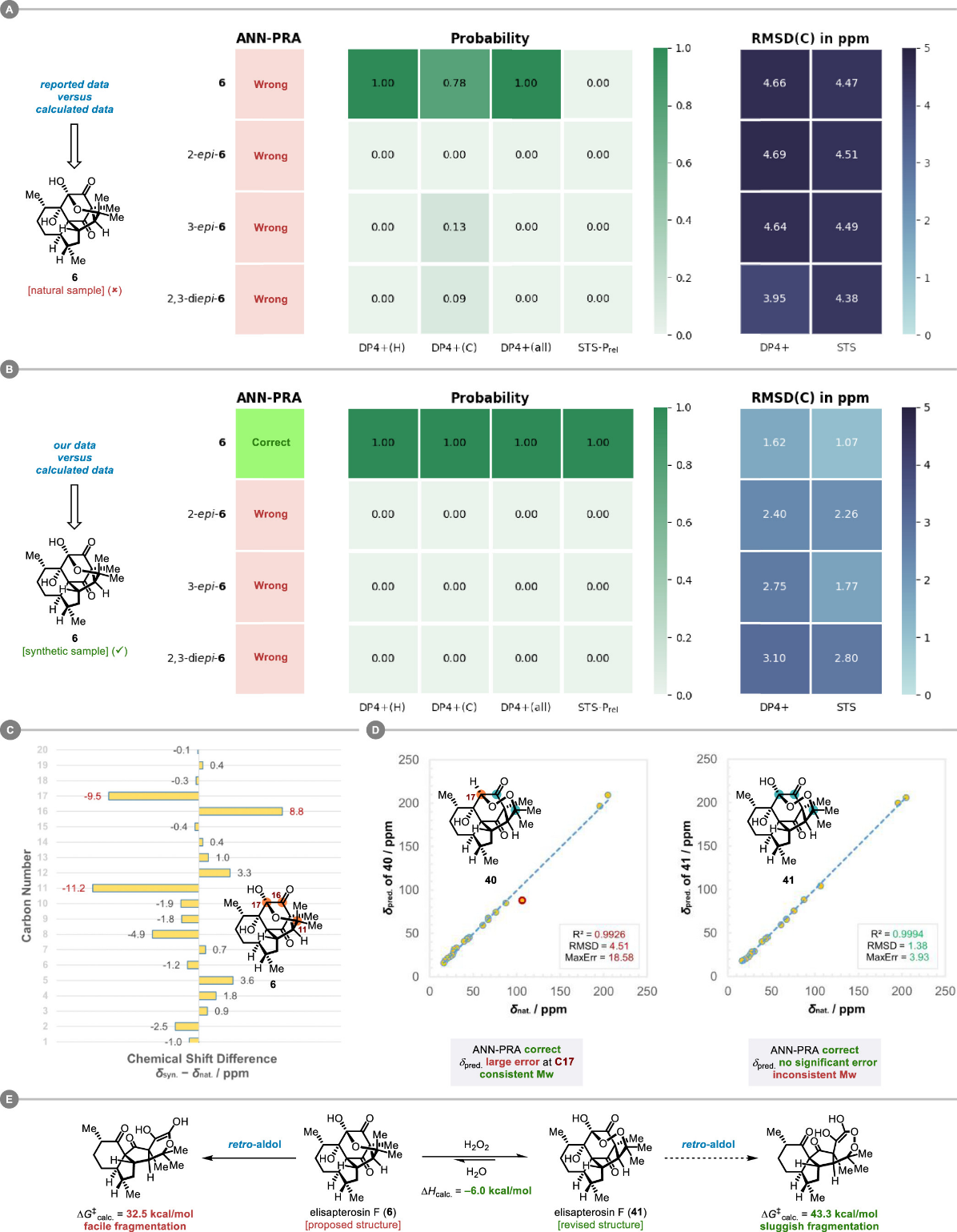
图九 图片来源:J. Am. Chem. Soc.
根据计算修正后的结构,研究团队首要任务是对elisapterosins B和C进行烯醇氧化条件的筛选,以期实现β-立体选择性控制。幸运的是,他们发现苯亚硒酸酐可作为有效的氧化试剂,并由此完成了elisapterosins A和D的合成(图十)。另一方面,借助于在研究过程中偶然发现的两个由分子内氢原子转移(HAT)介导的C17位羟基氧化途径,团队顺利实现了elisapterosins E和F的全合成(图十一)。至此,计算化学所预见的特殊结构变异成功指引了合成路线的设计,并最终获得实验结果的充分支持。研究团队也对通过类比和比较的方式推测有机化合物结构的做法提出了警示,因其不可避免带有研究者的主观判断。
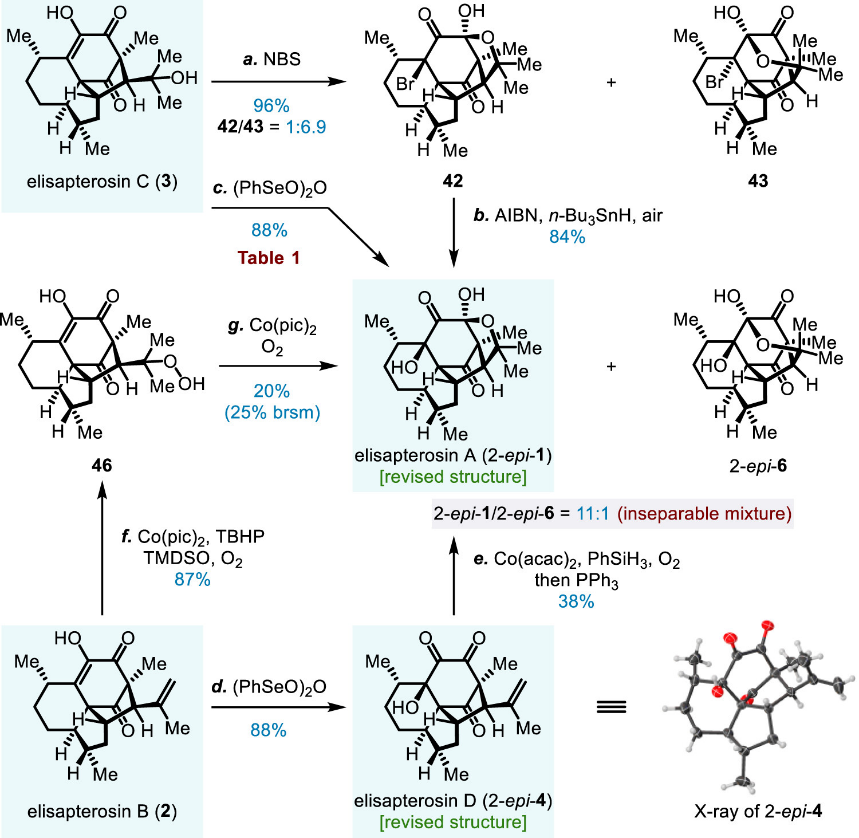
图十 图片来源:J. Am. Chem. Soc.
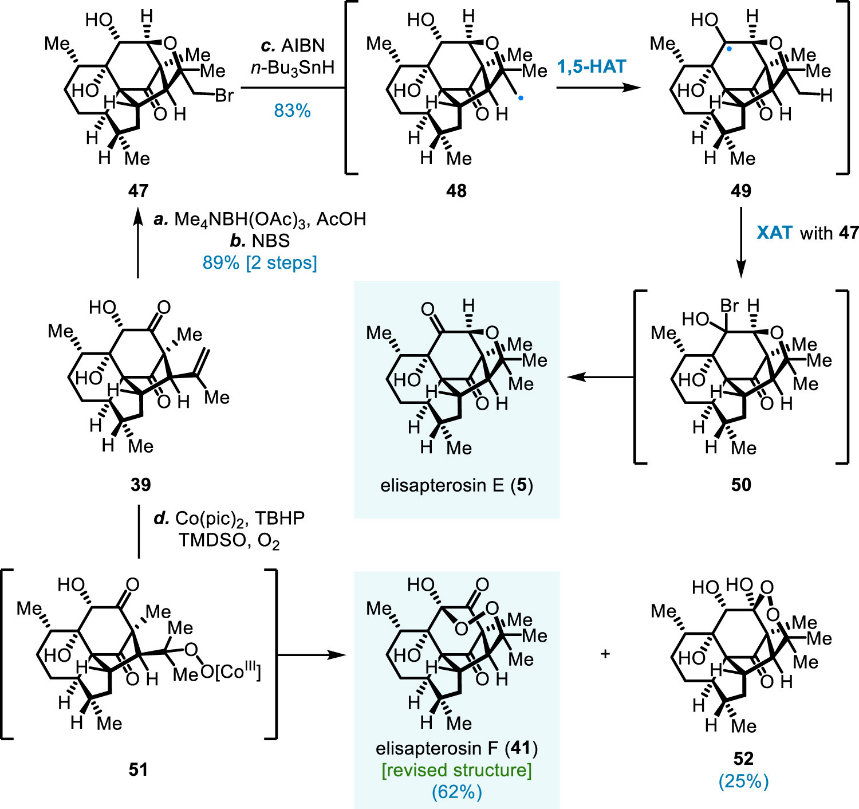
图十一 图片来源:J. Am. Chem. Soc.
该研究在丁寒锋教授指导下完成;浙江大学化学系博士生刘春慧及硕士生龚涵为该论文共同第一作者,完成了主要的实验工作;浙江大学化学系博士生夏启东为论文的共同通讯作者,完成了天然产物结构修正等理论计算工作。本研究得到了国家自然科学基金的资助。谨以此研究致浙江大学化学系成立110周年。
原文链接:https://pubs.acs.org/doi/10.1021/jacs.5c11010
丁寒锋课题组主页:
https://person.zju.edu.cn/ding
文字:丁寒锋教授课题组
编辑:黄珍珍、邹尔纯
审核:陆展