
史炳锋和洪鑫课题组:Cp*Co(III)催化的碳氢键活化/简单烯烃的不对称氢芳基化反应
(杂)芳基碳氢键对烯烃的立体选择性插入是获取含有苄位手性中心化合物的快捷办法。然而,为了得到较高的立体选择性,该类反应所采用的烯烃多局限于苯乙烯类、张力环状烯烃类(如降冰片烯)、电子失衡的活化烯烃类(如丙烯酸酯),或者局限于分子内的反应。迄今为止,以简单的、非活化的末端烯烃为原料的碳氢键键活化/不对称氢芳基化反应仅有少数成功案例。近日,我系史炳锋教授课题组和洪鑫课题组合作,借助第9族廉价过渡金属Co(III)催化的碳氢键活化策略,实现了简单末端烯烃高立体选择性的氢芳基化反应,并对反应机理进行了深入的研究(图1)。
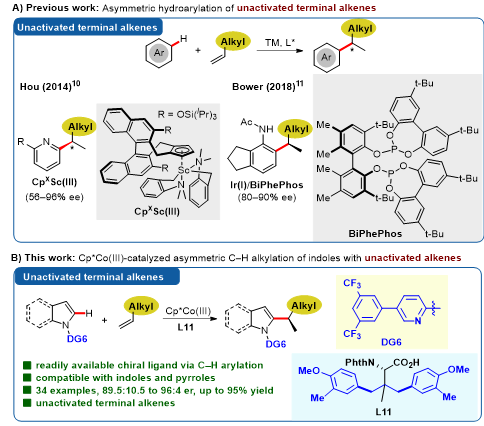
图1. 研究背景与本文工作
研究伊始,作者选用无取代吡啶为导向基团(DG1)、以1-辛烯为模型底物对基于氨基酸的手性羧酸配体进行筛选(图2)。结果发现产物3的er值随氨基酸侧链位阻变大而升高(L1, L0, L2),尤其是当侧链为叔丁基(L3)和金刚烷基(L4)时,产物er值有较大幅度的提高。
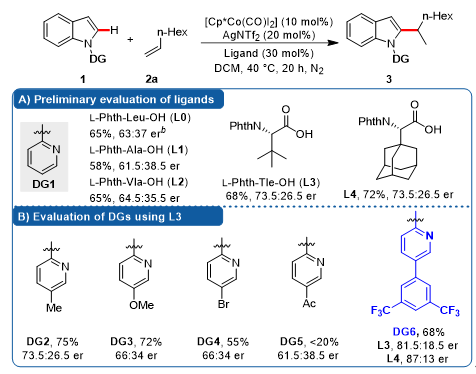
图2. 对手性配体及导向基团的初步考察
考虑到导向基团(DG)在反应中作为配体,不仅显著影响着中心金属Co(III)的电子立体效应,也对周围的手性环境产生影响,作者进一步考察了导向基团这一“配体”的变化对产物er值的影响。作者发现,在吡啶氮原子的间位引入的3,5-双三氟甲基取代的苯环取代基(DG6)可以大幅提升产物的er值(从73.5:26.5提升至81.5:18.5)。更有趣的是,当使用位阻更大的L4时,效果更佳(87:13 er)。至此,作者似乎找到了解决问题的方向,那就是借助导向基团DG6的优势,找到一种位阻更大并且(或者)与DG6有相互协同作用的手性氨基酸配体,来协同提高手性诱导。
鉴于配体L4难于改造,作者计划对廉价易得的叔亮氨酸L3中的三个甲基进行空间位阻扩充。采用课题组之前发展的Pd(II)催化叔亮氨酸γ-甲基碳氢键芳基化反应(Chem. Sci. 2020, 11, 290–294),作者快捷地制备了系列大位阻手性叔亮氨酸的衍生物,并对反应进行适配(图3)。结果显示双芳基化氨基酸配体L11可以顺利的将目标产物的er值提高至较高水平(95:5) (图3A)。值得注意的是,配体L11可以由上述制备方法一步制备(图3B)。
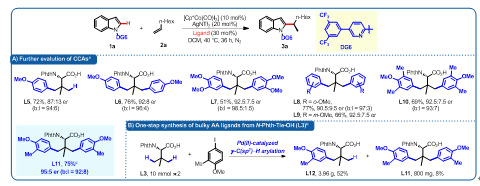
图3. 手性配体的合成和进一步考察
在底物的适用性上,简单末端烯烃与吲哚类底物反应,几乎都能均一地得到95:5以上的er值(图4A, B);对于吡咯类底物的手性诱导能力有所下降(图4C);该反应可以适用于分子内七元环化过程,并且er值不受影响(图4D);遗憾的是,反应对芳基类底物适用性较为有限(图4E)。
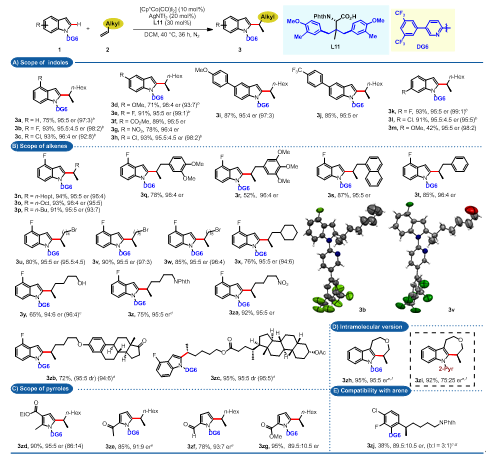
图4.底物考察
作者接着通过理论计算阐明了该不对称反应的机理(图5):1)碳氢键活化之后手性羧酸与有机Co(III)中间体中的导向基团DG6之间存在重要的π-π相互作用(中间态9);2) 手性羧酸配体的参与,有利于烯烃插入过程(9®11);3)烯烃插入是不可逆的,同时也是整个反应的手性决定步和决速步,这一结论也通过天然丰度13C KIE实验得以验证(图6);4)配位后的简单烯烃与配体中芳环的碳氢…π非共价相互作用(NCI)是手性引发的决定性因素(TS10-R)。
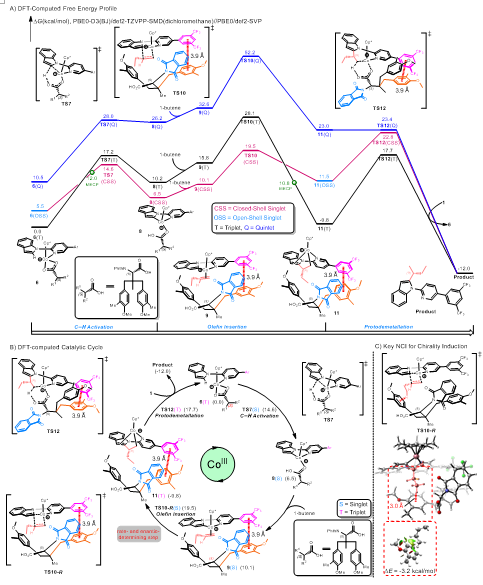
图5. DFT反应势能图及催化循环图
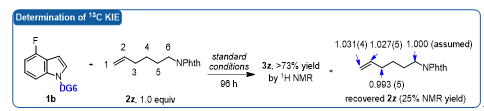
图6. 天然丰度13C KIE实验
综上,史炳锋和洪鑫课题组发展了一种非手性Cp*Co(III)和手性酸催化的碳氢键活化/非活化烯烃不对称氢芳基化反应。反应具有广泛的底物适用性,机理研究表明π-π堆积和碳氢…π非共价相互作用对反应的立体选择性具有显著影响。
论文信息:
Cp*Co(III)-Catalyzed Enantioselective Hydroarylation of Unactivated Terminal Alkenes via C-H Activation
J. Am. Chem. Soc. DOI: 10.1021/jacs.1c08562.