
亚胺化学键是不是真的“怕水”?浙大化学系师生说“未必”
利用可逆的亚胺缩合反应进行复杂分子的自组装,是动态共价化学的研究热点之一。亚胺化学键是醛与胺缩合并释放水分子而形成。根据勒夏特列原理,水环境下平衡向亚胺水解的方向进行。因此目前基于亚胺的缩合反应一般在油溶剂的条件下进行,其中一些体系使用移除水分子(如分子筛吸水、油水分离器等)来保证亚胺缩合反应在无水的条件下进行。换句话说,在水溶液下进行亚胺缩合自组装一般被认为极具挑战。
对于超分子科学的研究而言,水经常被认为是近乎完美的溶剂。一方面,水无毒无害,是生命介质,实现自组装分子的水兼容性十分重要。另一方面,在水溶液中,疏水作用能驱动主客体识别。因此,很多研究组开始思考解决亚胺和水不能兼容的问题。
李昊研究组在亚胺缩合反应过程中发现了一个“多重共价键加强稳定性”的现象,即,尽管单个的亚胺键在水环境下是不稳定的,当构成自组装体系的构筑基元之间有多个亚胺共价键联接在一起时,它们的稳定性相较单个亚胺键大大加强。这个现象在无机化学领域被称为“螯合效应”。

基于这个原理,浙江大学化学系李昊课题组在水溶液中进行亚胺缩合反应,实现了具有三维复杂拓扑结构的分子的高效自组装。如下图所示,当我们将柔性三胺分子TREN和含有羧酸根的三醛分子在水中混合时,在有有机模板分子(如氯仿或二氯甲烷)的情况下,一个四面体分子能以4+4的方式高效合成。
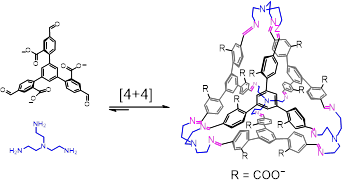
值得注意的是,如果将羧酸根移动到醛基的邻位,自组装的路径将完全不同。一个三棱柱分子将被自组装,如下图所示。
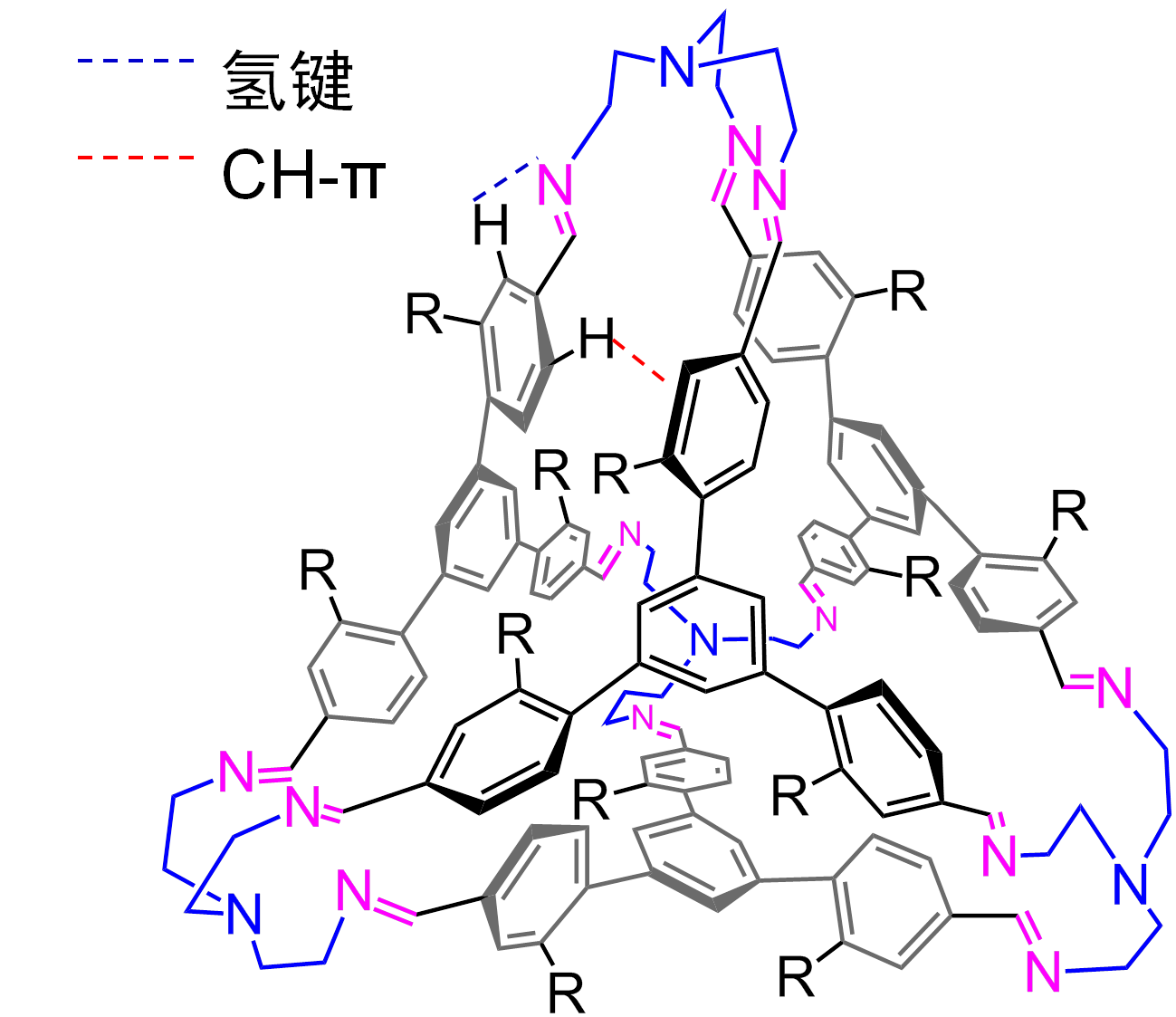
造成这个巨大的差异的原因是四面体分子的形成是依靠分子内驱动力的。如下图(四面体的一个片段)所示,四面体的形成依赖于亚胺邻位的两个质子参与CH-π作用和CH-N氢键。任何一个质子被羧基或其他基团取代,四面体都将失去形成的内在驱动力。
正是因为这个四面体分子和水溶液是兼容的,我们可以依靠疏水作用,让四面体分子识别一系列尺寸匹配的客体分子,如四氯化碳,环己烷等。为今后研究限域空间的超分子化学奠定了基础。
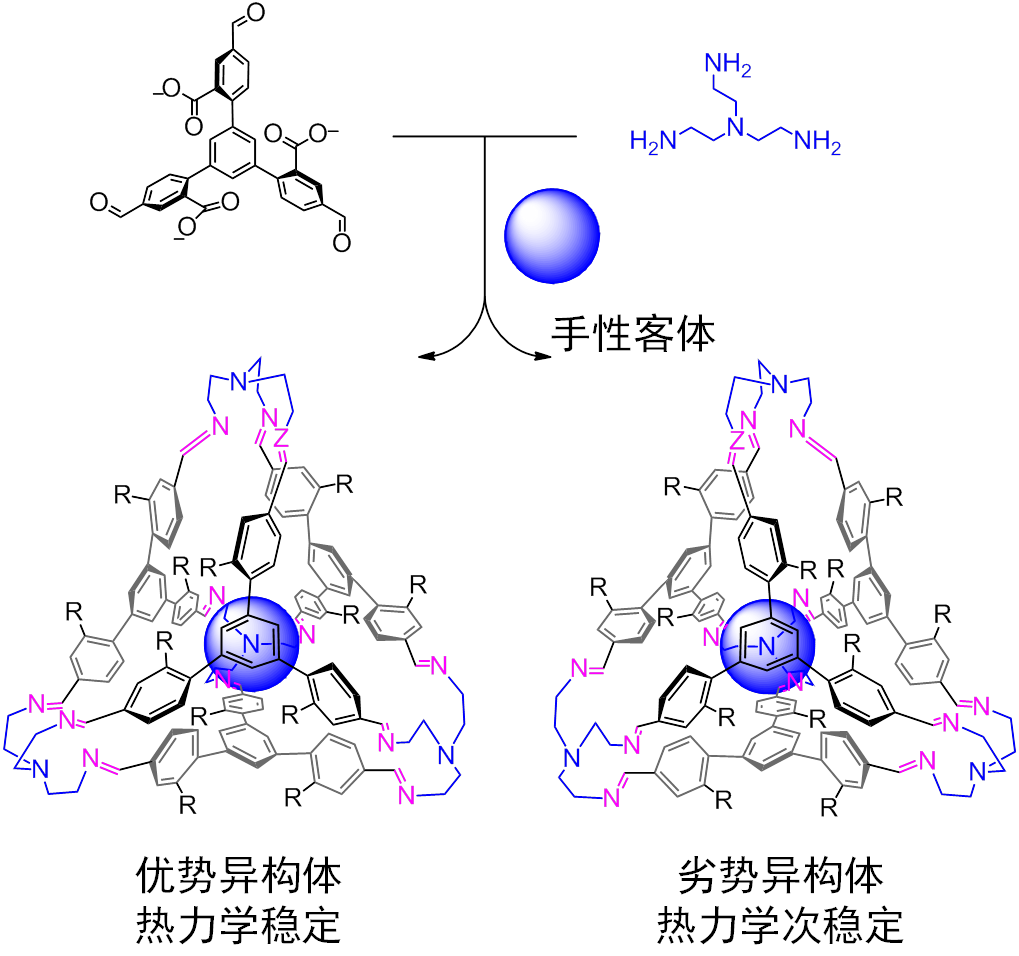
另一方面,由于分子内的CH-π作用,单个的四面体分子从而具有了手性。即构成四面体的四个角落的三个苯环具有统一的芳香性。笼状分子本质上是一对对映异构体。手性色谱实验表明,这一对对映异构体其实是可以拆分的。被拆分后的手性笼状分子的圆二色谱在一周的时间内没有衰减,证明了分子手性的稳定性。
而如果在体系中加入手性客体,由于笼状分子的两个对映异构体对手性分子识别的差异,其中一个对映异构体能被选择性的生成。圆二色谱证明笼子的ee值大约在10%左右。未来的研究将致力于利用笼子的手性环境,完成客体的手性诱导反应和手性分离。
相关工作发表于Angew. Chem. Int. Ed. DOI: 10.1002/anie.202106428R1。这个工作的一作是浙江大学化学系硕士研究生陈依昕。本工作得到了本系王琦教授,曾秀琼教授,厦门大学曹晓宇教授等老师的支持和指导。
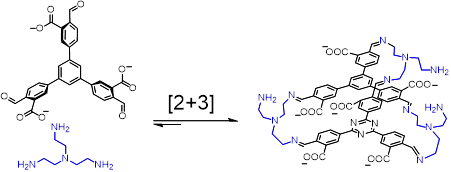
而稍早,同样利用“多重共价键”的策略,李昊研究组在纯水溶液中将一个六醛分子和手性二胺分子缩合,获得了一个手性的杯状分子。并利用该杯状分子的手性,实现了主体分子对手性客体的选择性识别。
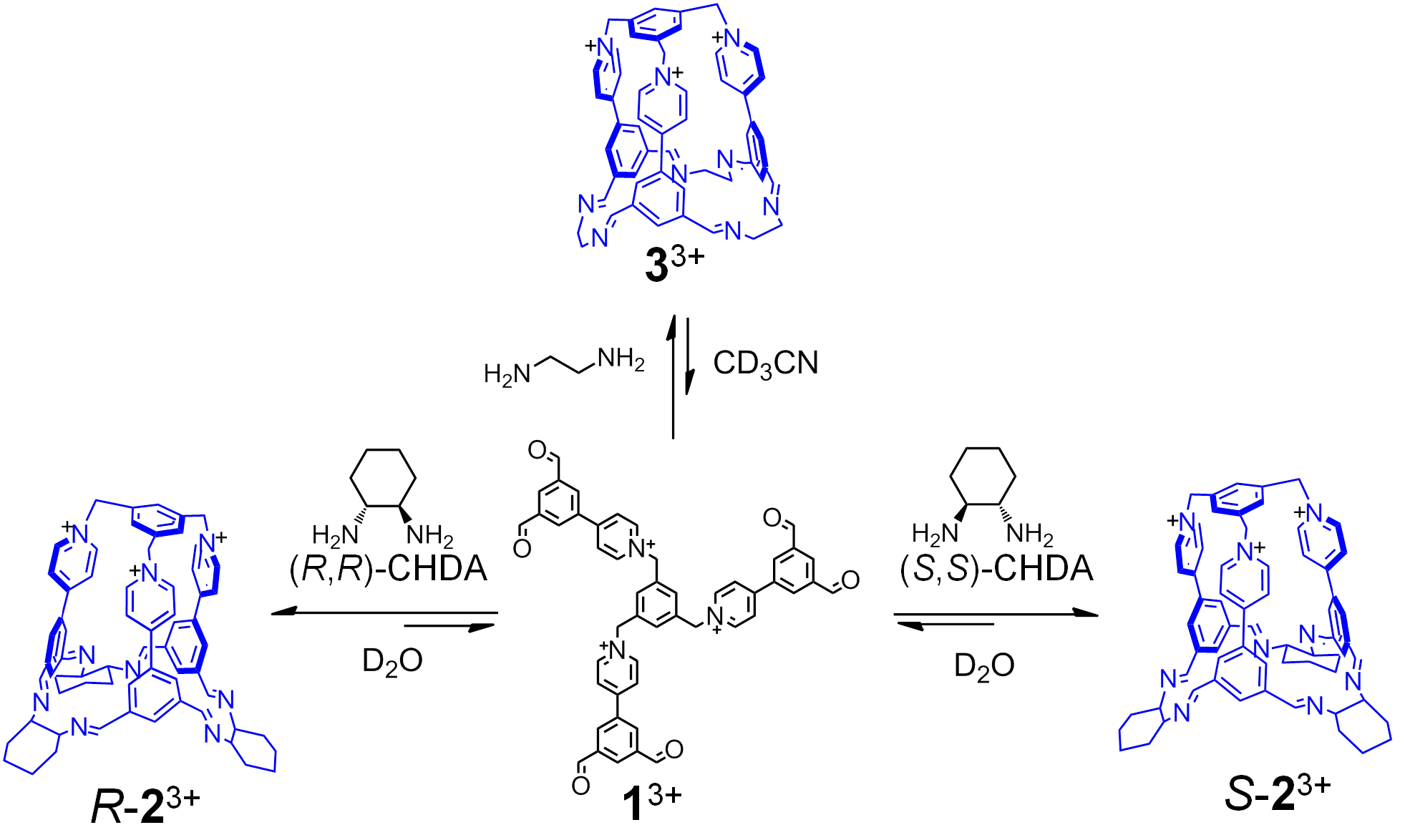
自组装是通过在水溶液中混合两种构筑基元完成的。其中13+含有六个醛基,反式环己烷二胺含有两个位于gauche位置的胺基。通过调节13+的反离子可以控制其水溶性和油溶性。在水溶液中,笼状分子23+能以60%的产率组装。如果将溶液换成乙腈,由于乙腈分子的模板效应,产率则可以接近定量。反式环己烷二胺的手性使笼状分子23+具有手性空腔,RR和SS的环己烷二胺与13+分别组装成R-23+和S-23+;将外消旋的反式环己烷二胺与13+混合,体系则会发生自分类现象,生成R-23+和S-23+的外消旋体。这些手性笼在稀释、离子交换沉淀以及加入竞争性的醛或胺后,均没有明显的亚胺副分解反应和水解反应。
当用乙二胺代替反式环己烷二胺时,在水溶液中的自组装并没有后者那么成功,而是生成了大量的聚合物。这是因为相比于环己烷二胺,乙二胺具有更高的柔性,自组装形成的产物会损失更多的熵。幸运的是,在乙腈中,乙二胺和13+能自组装成R-23+和S-23+的非手性类似物33+。一旦组装成功并交换离子后,这些笼状分子就具有了优秀的动力学惰性,能在水相中稳定存在一个月。

由于R-23+和S-23+具有手性空腔,能在水中对手性客体进行识别。研究表明,S-23+能选择性识别R构型的1,2-环氧丁烷,而对S构型的客体几乎没有识别能力。
相关研究结果发表于Angew. Chem. Int. Ed. 2021, 60, 4705。该工作的一作是浙江大学化学系的博士研究生雷晔,该工作得到了本系潘远江教授,黄飞鹤教授等老师的支持和指导。