
丁寒锋/宣军团队JACS:Hetidine型C20-二萜生物碱的发散式不对称全合成
近日,我系丁寒锋/宣军课题组在Hetidine型C20-二萜生物碱的全合成研究中取得重要进展,相关成果以“Asymmetric Total Syntheses of Hetidine-Type C20-Diterpenoid Alkaloids: Spirasines V and VI, Spiradine D, and the Proposed Structures of Spirafines II and III”为题,发表于《美国化学会志》(JACS)(https://doi.org/10.1021/jacs.5c13564)。研究团队发展了一种汇聚式不对称合成策略,以17–20步反应高效、高立体选择性地完成了五个Hetidine型C20-二萜生物碱(spirasines V和VI、spiradine D,以及spirafines II和III推测结构)的全合成。关键反应包括Enders不对称加成、氧化去芳香化促进(ODI)的Diels–Alder环加成,以及金属氢化物氢原子转移(MHAT)介导的跨环自由基环化,并通过后期还原环化策略构建杂环体系。
C20-二萜生物碱主要分离自乌头属、翠雀属、飞燕草属及绣线菊属植物,具有结构多样性和显著的生物活性,在传统药物中沿用数百年。Hetidine型生物碱作为生源上连接Atisine型与Hetisine型的中间体,具有独特的笼状六环骨架。然而,由于合成难度大,该亚类生物碱的全合成研究较为有限,此前仅马大为、李昂、张敏等课题组完成了少数Hetidine型生物碱的全合成(图1)。

图1.此前已实现全合成的Hetidine型生物碱(图片来源:J. Am. Chem. Soc.)
本文研究的五个天然产物均来源于绣线菊属植物。其中,spiradine D表现出优异的抗血小板活化因子活性,但其天然来源稀缺,限制了深入生物学研究。为实现该类分子的高效制备,课题组提出了一种模块化合成策略。逆合成分析显示,五环中间体11可作为合成关键节点,通过还原胺化引入哌啶环,获得spirafines II(9)和III(10)。进一步,spirafine III可通过还原环化构建噁唑烷环,从而发散性合成其余目标分子。中间体11由MHAT介导的跨环自由基环化构筑关键的C9–C10键;其前体13则通过ODI-Diels–Alder反应高效构建;而C5位手性中心则经由Enders不对称加成高对映选择性引入,为整体立体化学控制奠定基础(图2)。
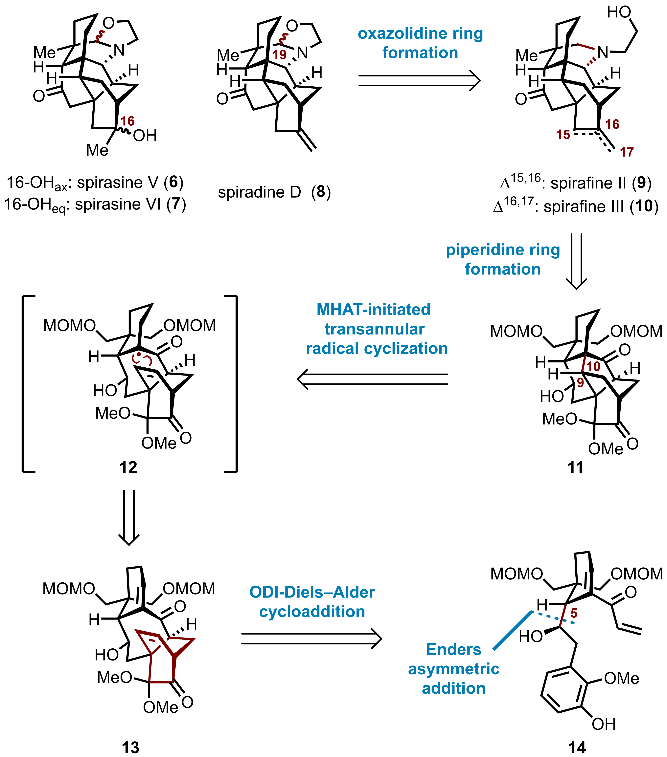
图2.逆合成分析(图片来源:J. Am. Chem. Soc.)
从带有手性辅基的原料15出发,与醛16经Enders不对称加成制得化合物17,随后通过臭氧化/Dess–Martin氧化、共轭还原、三氟甲磺酸酯化及酮的立体选择性还原等步骤,获得高对映选择性(96% ee)的烯醇三氟甲磺酸酯19。进一步经羰基化/内酯化、乙烯基开环/脱保护等转化,得到Diels–Alder前体14。在醋酸碘苯(PIDA)作用下,通过ODI-Diels–Alder环加成专一性得到13,进而经MHAT介导的跨环自由基环化高效构建核心笼状五环骨架11(图3)。
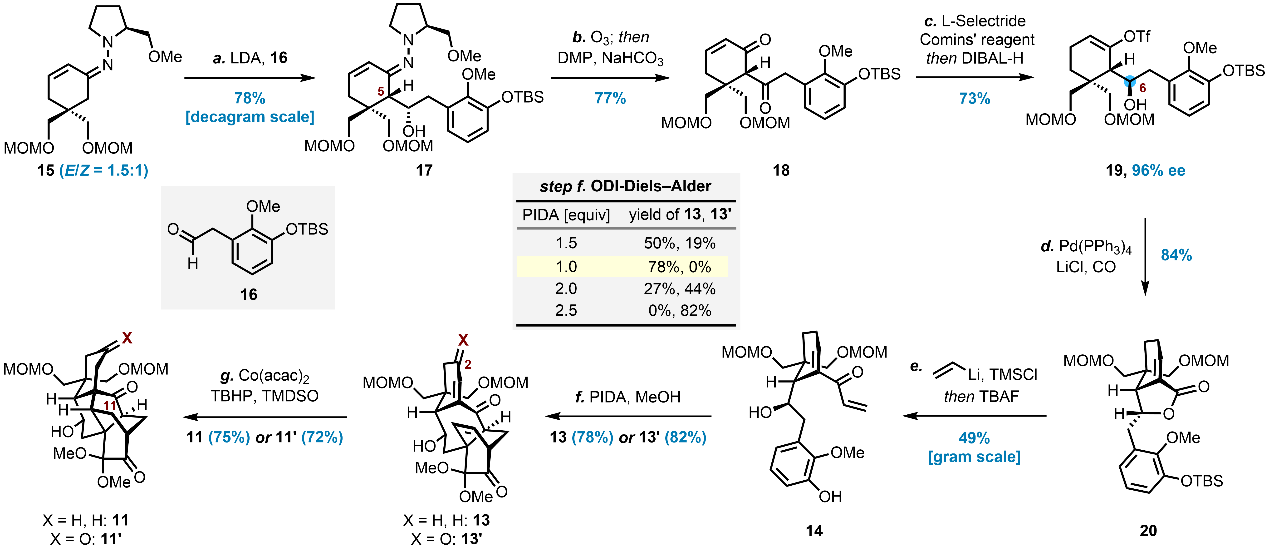
图3. 关键五环中间体的合成(图片来源:J. Am. Chem. Soc.)
研究进一步发现,C6位羟基的立体构型对MHAT环化反应至关重要(图4)。由差向异构体19衍生的前体22在相同条件下未发生预期环化,而是经历氢化过氧化/半缩酮化驱动的Grob型碎片化,生成副产物24。此外,乙酰基保护中间体21仅得到氢过氧化及还原产物。理论计算表明,两类底物的过渡态TS1与TS2之间存在4.4 kcal/mol的能量差,源于环化过程中原子间距离缩短导致的范德华斥力增强。
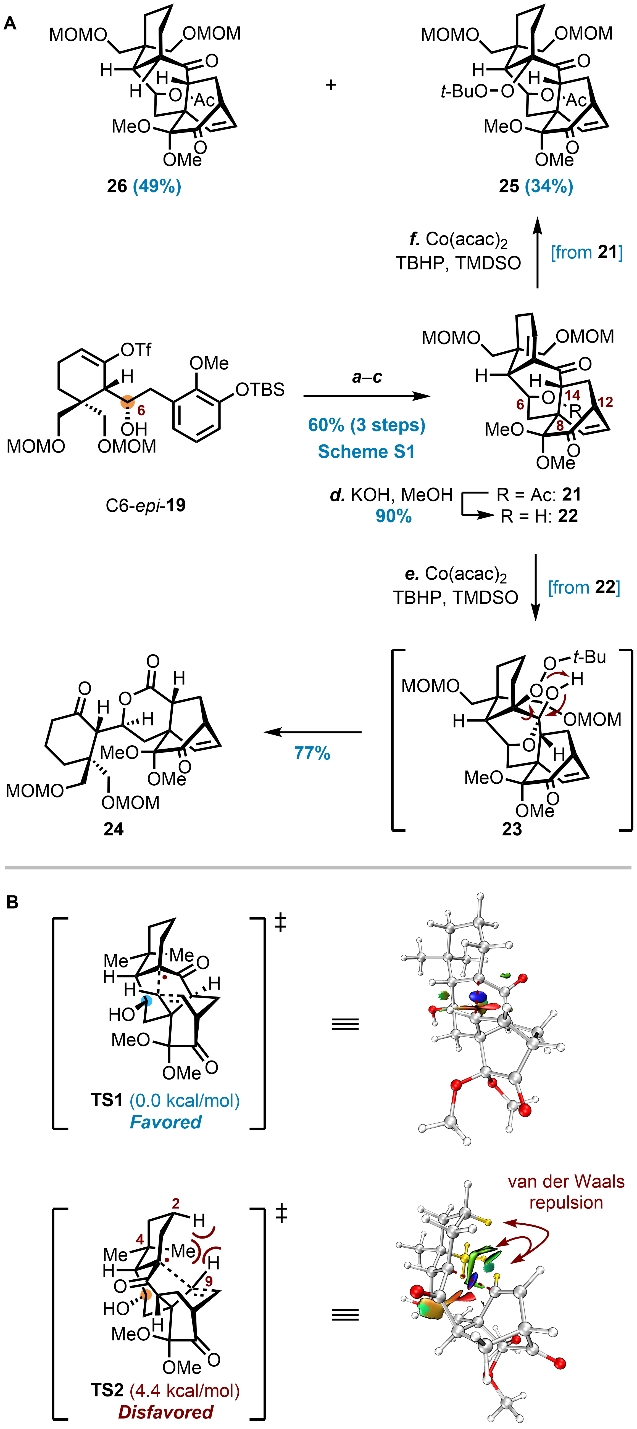
图4. MHAT介导的跨环自由基环化反应研究(图片来源:J. Am. Chem. Soc.)
从中间体11出发,经还原脱甲氧基化、Petasis亚甲基化及脱MOM保护制得三醇27。利用位阻控制实现硫代碳酸酯选择性构建,再经自由基脱氧与PDC氧化得酮醛29。随后通过还原胺化构建哌啶环、Ley–Griffith氧化引入羰基、碱促C6–N键断裂等步骤,完成spirafine III(10)推测结构的全合成(图5)。RhCl3催化烯烃异构化可将其转化为spirafine II(9)推测结构。尽管二者结构经单晶衍射确认,其核磁数据与文献报道存在差异。进一步,通过Mukaiyama水合在C16位引入叔醇。值得注意的是,中间体32在Ley–Griffith氧化条件下生成内酰胺34,推测经由[1,4]-氢迁移/氧化过程在C19位引入羰基。最终,经构建噁唑烷环完成spirasine V(6)的全合成,获得一对C19差向异构体(1.2:1 dr)。相同路线亦可得spirasine VI(7),其与HCl成盐后均转化为单一异构体,核磁数据与文献一致,且7的19S构型经单晶确认。最终,7经脱水转化为spiradine D(8)。
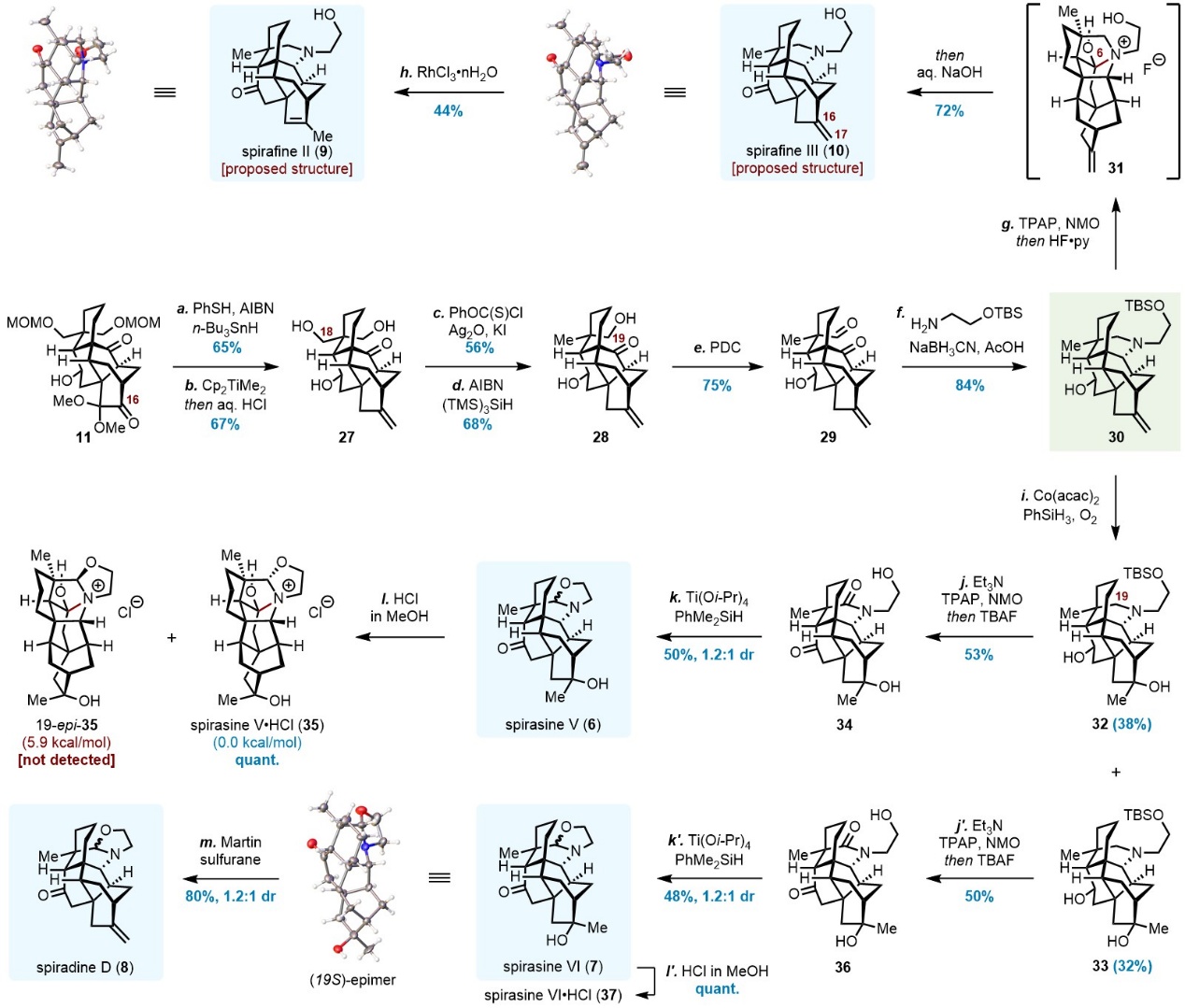
图5. Spirasines V和VI,spiradine D以及spirafines II和III推测结构的全合成(图片来源:J. Am. Chem. Soc.)
总结
丁寒锋/宣军团队通过Enders不对称加成、ODI-Diels–Alder环加成与MHAT跨环自由基环化等关键反应的协同运用,高效构建了Hetidine型C20-二萜生物碱的核心骨架,并在合成后期精准引入杂环结构,首次实现了五个目标分子的不对称全合成,为后续功能研究奠定基础。该合成策略中采用的C9–C10断键思路显著简化了路线设计,为含不同氧化态C20-二萜生物碱的合成提供了通用、模块化的平台。
本文第一作者为浙江大学化学系姚凤洁博士,化学系本科生盛伊甸同学完成了理论计算工作,丁寒锋教授和宣军研究员为共同通讯作者。本研究得到了浙江大学化学系和国家自然科学基金的支持和资助。
论文链接:https://pubs.acs.org/doi/10.1021/jacs.5c13564
课题组介绍
丁寒锋/宣军团队主页:
https://person.zju.edu.cn/ding
https://person.zju.edu.cn/xuanjun
图文:丁寒锋教授课题组
编辑:黄珍珍、邹尔纯
审核:陆展