
史炳锋教授团队ACS Catalysis: Pd(II)催化的对映选择性C(sp3)–H芳基化合成膦中心手性二烷基膦酰胺

导语:
膦中心手性化合物在手性配体、有机小分子催化剂以及生物分子等领域中发挥着重要作用,因此,发展高效的不对称催化方法构建膦中心手性分子受到化学家们的广泛关注。在已经报道的方法中,过渡金属催化的不对称碳氢键活化策略构建膦中心手性的分子合成中表现出了广阔的应用前景。然而截至目前,已有的报道均局限于芳基膦化合物的对映选择性C(sp2)–H活化合成芳基取代的膦中心手性化合物。
近期,浙江大学史炳锋教授团队通过偕二乙基去对称化策略实现了首例不对称C(sp3)–H活化合成膦中心手性二烷基膦酰胺化合物。作者利用他们课题组发展的2-吡啶基异丙基酰胺(PIP)作为导向基以及简单3,3'-CN2-H8-BINOL作为手性配体,成功以中等到良好的收率和优秀的对映选择性合成了一系列手性二烷基膦酰胺。为大位阻膦中心手性化合物的合成提供了新的思路。相关工作发在ACS Catalysis (DOI: 10.1021/acscatal.4c01212)
前沿科研成果:Pd(II)催化的对映选择性碳氢键活化合成膦中心手性二烷基膦酰胺
过渡金属催化的不对称碳氢键活化已经发展成为一种行之有效的构建各类手性骨架的策略。相较于已经取得显著发展的不对称sp2碳氢键活化,sp3碳氢键因为其低酸度、高解离能,以及缺少可以与金属催化剂相互作用的低能级空轨道等特点,其不对称活化一直以来都更具挑战性。根据新生成的手性中心与碳-金属键的距离,不对称sp3碳氢键活化大体可分为三种策略(图1,a)。第一种是亚甲基不对称碳氢键活化,对同一个碳上两个氢原子进行区分,在这种策略中新生成的手性中心直接与金属原子相连(图1,a,Pathway I);第二种途是偕二甲基去对称化,这种情况下新生成的手性中心与碳-金属键处于相邻位置(图1,a,Pathway II);第三种途径中是通过对更长的烷基链进行去对称化,新生成的手性中心远离金属催化剂(超过两个键的距离)(图1,a,Pathway III)。其中,第三种途径因为手性配体与手性中心相距远、反应过程中底物构象更加灵活多变,其立体控制具有很强的挑战性。截至目前,只有余金权课题组成功报道了一例通过偕二乙基的去对称化构建β-碳中心手性。另外,已经报道的不对称sp3碳氢键活化都集中在构建碳中心手性的研究。

图1 研究背景
自2015年以来,过渡金属催化的不对称碳氢键活化构建膦中心手性分子受到了广泛关注,然而这些工作均局限于芳基膦化合物的对映选择性C(sp2)–H活化(图1,b)。另一方面,烷基取代的膦中心手性化合物得益于它们独特的强供电子能力以及大空间位阻,已经被证明是不对称催化中非常有效的手性催化剂(如BisP*, QuinoxP*, BIBOP, TangPhos等,图1,c)。然而目前合成这类化合物的高效的不对称合成方法鲜有报道。史炳锋教授团队长期致力于惰性碳氢键的精准催化转化研究,为了解决非活化亚甲基碳氢键的不对称活化的难题,该团队发展了具有偕二甲基效应的2-吡啶基异丙胺(PIP-NH2)导向基试剂,实现了惰性亚甲基sp3碳氢键的高效官能团化(Angew. Chem. Int. Ed.2013, 52, 13588; Chem. Sci. 2013, 4, 4187; J. Am. Chem. Soc. 2015, 137, 8219; Acc. Chem. Res.2021, 54, 2750)。该课题成功地利用PIP导向基中偕二甲基与联萘骨架配体的“位阻传递(steric communication)”效应,实现了非活化亚甲基碳氢键的不对称碳氢键活化(Angew. Chem. Int. Ed.2018, 57, 9093;J. Am. Chem. Soc. 2019, 141, 4558;Chin. J. Chem.2020, 38, 242;Angew. Chem. Int. Ed.2020, 59, 14060;Angew. Chem. Int. Ed.2020, 59, 20455)。基于以上研究基础,他们认为可以通过PIP导向不对称sp3碳氢键活化/偕二乙基去对称化策略,直接高效地构建手续二烷基膦中心手性化合物(图1,d)。
作者使用PIP衍生的二乙基膦酰胺1与4-碘苯甲醚作为标准底物筛选反应条件(表 1)。他们发现在使用PdI2作催化剂,(S)-BINOL作配体,碳酸钾作为碱的情况下,在氮气氛围中120度反应12小时可以以22%的产率、18%的ee值收获目标产物。通过对其他类型BINOL配体的筛选,作者发现,在3,3’位引入氰基能显著提升反应的收率及对映选择性。然而在反应性提高的同时,双芳基化的副产物产率也显著提高,使得目标产物3a的产率难以得到进一步提升。随后的配体筛选中作者发现3,3'-CN2-H8-BINOL能很好的抑制副产物的生成,通过减少配体的量以及延长反应时间到18小时,最终以77%的分离收率,99%的ee值得到目标产物。
表1 反应条件优化
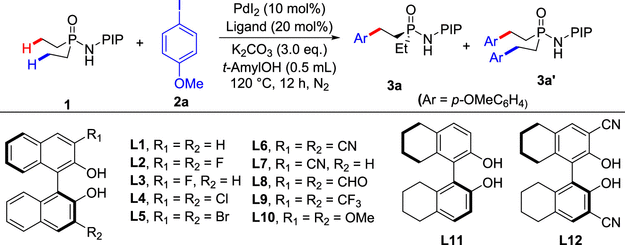
entry | ligand | yield of 3a (%) | yield of 3a’ (%) | ee (%) |
1 | L1 | 22 | trace | 18 |
2 | L2 | 16 | trace | 91 |
3 | L3 | trace | none | - |
4 | L4 | 20 | trace | 18 |
5 | L5 | 17 | trace | 31 |
6 | L6 | 44 | 12 | 96 |
7 | L7 | 9 | none | 39 |
8 | L8 | trace | none | - |
9 | L9 | trace | none | - |
10 | L10 | trace | none | - |
11b | L6 | 37 | 16 | 94 |
12c | L6 | 42 | 15 | 96 |
13 | L11 | 5 | none | 63 |
14 | L12 | 44 | trace | 99 |
15d | L12 | 72 | trace | 99 |
16c, d | L12 | 81(77)e | trace | 99 |
在确定了最优反应条件后,作者考察了一系列芳基碘化合物在反应中的耐受性(图2)。总得来说,所考察的芳基碘化合物几乎都可以以优秀的对映选择性,中等到良好的收率得到对应的芳基化产物。取代基的电子效应对反应的收率有着较为显著的影响,吸电子取代基会导致产物收率降低。值得注意的是,此方法同样兼容包括吡啶、吲哚、苯并呋喃,苯并噻吩在内的几种杂环骨架的芳基碘化合物。另外,一些特殊的分子,例如荧光分子和天然分子衍生的芳基碘化合物也适用于该反应。该方法也适用于动力学拆分,作者对甲基乙基的膦酰胺底物(rac)-4的进行拆分,最终以28%的产率和95%的ee值得到了目标产物5a(图3)。
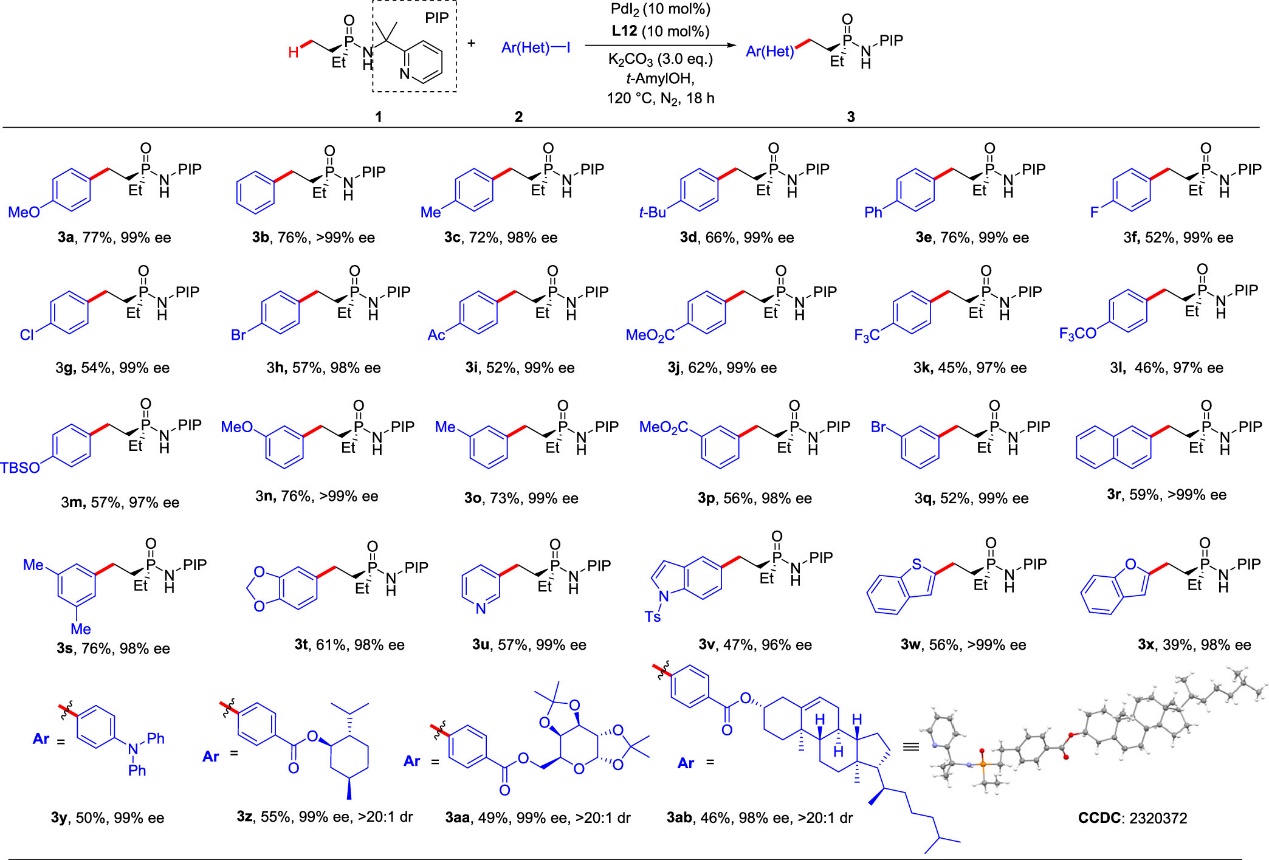
图2 芳基碘化合物普适性研究
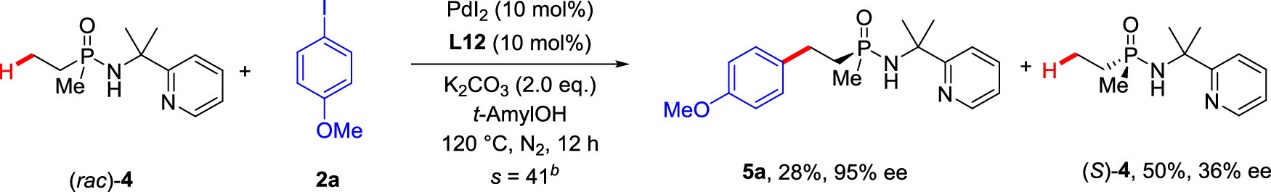
图3动力学拆分
为了展示该方法在合成上的应用潜力,作者进行了克级规模实验以及对产物进行了后续的转化实验(图4)。他们将标准反应扩大到4.0 mmol,仍可以以68%的产率以97%的ee值,得到0.94 g的目标产物3a。利用TfOH可以顺利实现导向基脱除,以63%的产率和95% ee得到二烷基膦氧化合物6a。随后,他们利用各类Grignard试剂对6a进行亲核取代,得到了多种三烃基取代的膦中心手性化合物7a-7c。最后,他们对三烷基膦化合物7a进行还原,以93% ee值得到了硼烷络合物的形式8a。

图4 合成应用
最后,他们还探究了配体L12的ee值与产物ee值的关系(图5),发现得到的是一条非线性的曲线,有较为明显的手性放大效应。说明在手性决定步中,可能有不止一个手性配体分子参与。
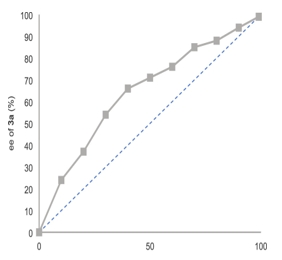
图5 非线性效应研究
综上所述,史炳锋教授团队报道了首例利用不对称C(sp3)–H活化策略构建膦中心手性二烷基膦酰胺化合物的方法。该工作以“Pd(II)-Catalyzed Enantioselective C(sp3)–H Arylation toward P-Stereogenic Dialkylphosphinamides”为题发表在ACS Catalysis(DOI: 10.1021/acscatal.4c01212)上。论文第一作者为2022级硕士生王辰跃,通讯作者为浙江大学史炳锋教授以及周涛副研究员。
史炳锋教授课题组主页:
https://person.zju.edu.cn/bfshi
文 字:史炳锋教授课题组
编 辑:黄珍珍、张维娅
审 核:林旭锋
终 审:丁立仲