
史炳锋教授姚启钧研究员团队JACS:钴催化不对称碳氢活化/[3+2]亲核环化反应构建手性桥双环化合物

手性桥双环骨架因其刚性的多环结构,被广泛应用于生物与材料领域。特别是手性[2.2.1]-桥双环结构,近年来,国内外多个课题组报道了其在不对称催化、药物合成与材料领域的重要应用突破。其中,桥杂双环烯烃,被广泛认为是多取代手性多环化合物的重要合成子。目前以桥杂双环烯烃为反应试剂,利用过渡金属催化不对称碳氢键活化构建手性分子的策略开始受到关注。但目前该策略仍高度依赖合成复杂、修饰困难的Cpx配位的贵价过渡金属。因此发展利用廉价、绿色的3d过渡金属与简单配体催化体系,实现对桥双环分子的不开环去对称化反应,是重要的研究方向。
浙江大学化学系史炳锋教授团队长期致力于发展绿色高效的廉价金属催化的不对称碳氢键活化新方法,自2019年以来,团队围绕廉价金属钴催化的不对称碳氢键活化,进行了系统而深入的探索。团队发展了大位阻手性酸配体(Chem. Sci.2020, 11, 290; J. Am. Chem. Soc. 2021, 143, 19112)和含有氢键受体酰胺基团的联萘/螺环手性酸(J. Am. Chem. Soc. 2021, 143, 6810; ACS Catal. 2022, 12, 9806),实现了非手性环戊二烯基钴[CpCo(III)]催化的不对称碳氢键活化。最近,史炳锋教授团队基于对金属钴配位模式的理解,提出了无Cp(cyclopentadienyl,环戊二烯基)配位的正八面体钴催化不对称碳氢键活化新策略,发展了两类新型手性催化体系,实现了廉价金属钴催化的不对称碳氢键活化。2021年,他们率先发展了Co(II)/螺环手性磷酸(SPA)接力催化体系,实现了螺中心手性的构建(Angew. Chem. Int. Ed. 2021, 60, 23187)。随后,他们又发展了新型Co(II)/Salox(salicyloxazoline,水杨基噁唑啉)催化体系,以廉价易得的二价钴盐作为催化剂和合成简便且便于改造的Salox手性配体,在体系中原位氧化生成正八面体手性Co(III)催化剂,高效地实现了不对称碳氢键活化/环化反应构建膦中心手性,并成功地分离和表征了相应的正八面体型Co(III)关键反应中间体(Angew. Chem. Int. Ed.2022, 61, e202202892)。随后他们将该催化模式成功应用于双轴手性化合物的构建(Angew. Chem. Int. Ed. 2022, 61, e202208912)、膦酰胺不对称碳氢键脱氢烷氧基化和胺化(Angew. Chem. Int. Ed. 2022, 61, e202210106; Angew. Chem. Int. Ed.2023, 62, e202302964)及首例对映和区域选择性电氧化钴催化C-H/N-H环化反应(Angew. Chem. Int. Ed. 2023, 62, e202218533)、手性二芳基甲胺的构建(Angew. Chem. Int. Ed.2023, 62, e202304706)、联芳轴手性骨架的构建(Angew. Chem. Int. Ed. 2023, 62, e202310004)、C1-手性四氢异喹啉(c-THIQs)生物活性分子的不对称合成(J. Am. Chem. Soc. 2023, 145, 24499)、不对称碳氢键活化/[4+1]插羰环化构建手性异吲哚啉酮骨架(Angew. Chem. Int. Ed. 2024, 63, e202318803)和不对称碳氢键酰氧化构建氧代二茂铁面手性化合物(CCS Chem. doi:10.31635/ccschem.024.202303709.)
受以上工作启发,该团队成功地实现了苯甲酰-8-氨基喹啉底物或N-2-吡啶芳基腙类底物的不对称碳氢键活化/[3+2]亲核环化反应,一步构建具有连续4-5个手性中心的手性[2,2,1]-桥双环化合物。相关成果近期在线发表于J. Am. Chem. Soc.上。
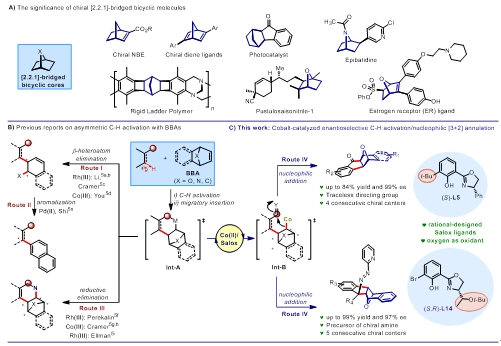
图1. 手性桥双环分子的应用与合成方法。图片来源:J. Am. Chem. Soc.
受课题组所发展的Co/Salox催化体系的启发,作者设计了如下不对称串联反应:Co催化碳氢键活化/桥杂双环烯烃不对称迁移插入生成的Int-B,随后发生Co(III)-C键分子内亲核进攻,合成一个具有四个连续手性中心的新桥双环化合物。然而,建立这样的不对称合成路线仍面临许多挑战,例如:1) 桥头原子为杂原子(例如,氧杂,氮杂)时,其具有离去的能力,且存在环张力,该反应更容易通过Int-B中间体发生β-杂原子消除,得到不对称开环产物;2) 即使β-杂原子消除被成功抑制,三价钴中间体还原消除生成的[4+2]环化产物与Co(III)-C键亲核进攻发生[3+2]环化之间也存在竞争。
为了初步探索该反应的可行性,作者使用8-氨基喹啉为导向基的苯甲酰胺底物1a与配体L1通过当量反应制备了碳氢键金属化中间体(RC)-Co-1。当(RC)-Co-1与1.0 eq的氧杂苯并降冰片烯2a反应时,能以60%的产率与76%ee制备预期的手性化合物3aa。这说明,在Co/Salox催化体系中,该反应在形成Co-C中间体后趋向发生分子内亲核进攻,而不是β-杂原子消除与C-N还原消除,符合研究预期。同时,通过(RC)-Co-1的单晶与包埋体积图,可以发现该中间体结构中烯烃配位的手性口袋呈现出“空旷”的平面,因此反应仅诱导出中等的对映体选择性。
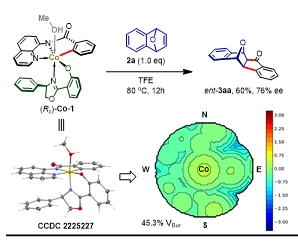
图2. (Rc)-Co-1的当量转化、单晶与包埋体积图。图片来源:J. Am. Chem. Soc.
为了对反应中间体存在的“手性口袋”进行修饰,作者对配体进行筛选。基于对C-Co中间体(RC)-Co-1的观察,作者重点筛选了在酚羟基邻位引入取代基的Salox配体。通过实验结果可以发现,随着在取代基体积的增大,反应的对映体选择性得到了显著提高。当使用邻位叔丁基取代的配体(S)-L5时,反应能够得到最佳的反应产率73%与对映体过量93% ee。
为了更好的了解反应的反应机制,作者对反应的机理进行了探究。作者使用1a、Co(acac)3、(S)-L5、二甲氨基吡啶(DMAP)在标准反应条件下以87%的产率制备了碳氢键活化后的Co-C中间体(Sc)-Co-2。通过对照实验发现,当反应中不加入碱LiOtBu时,反应不发生,因此碱的存在对于反应的碳氢键活化过程是至关重要的。当反应中不加入醛时,仍能以89%的产率制备(Sc)-Co-2,说明醛的添加并不影响反应碳氢键金属化过程。
作者使用制备的Co-C中间体(Sc)-Co-2与氧杂苯并降冰片烯2a进行当量转化实验。在标准反应条件下,能以81%产率,>99% ee得到3aa,更加验证(Sc)-Co-2为反应中间体,且此时能以78%的产率回收配体(S)-L5;而当反应中不添加醛或碱时都不能回收配体。作者推测碱与醛的加入促进了配体的释放,从而促进反应催化循环。受前人研究工作启发,反应中8-氨基喹啉作为原位脱除的导向基,抑制反应活性,因此推测醛的加入与8-氨基喹啉会生成亚胺并促进8-氨基喹啉与三价钴的配位解离,从而减小8-氨基喹啉对催化剂的毒化。
通过观察与(Sc)-Co-2同系的络合物(Sc)-Co-3单晶结构,作者发现(S)-L5噁唑啉侧链的苯环与底物8-氨基喹啉存在π…π堆叠作用,促进环金属中间体的生成。通过 (Sc)-Co-3包埋体积图,发现从(Sc)-Co-1到(Sc)-Co-3,由于邻位叔丁基取代基引入,中间体的包埋体积由45.3%增加至50.8%,使得喹啉基团呈现出轻微的扭曲阻挡了第二象限,同时阻碍桥双环烯烃2a从 “东半球”方向的进攻,限定2a的配位的取向,诱导得到优秀的对映体选择性。
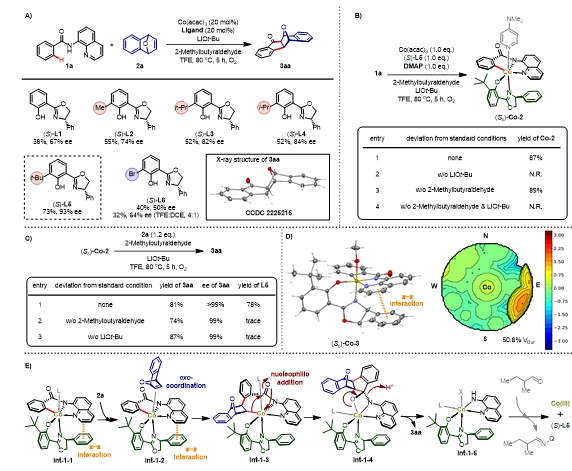
图3. 反应配体筛选与机理研究。图片来源:J. Am. Chem. Soc.
综合上述机理实验,作者提出一个可能的催化过程:三价钴盐与(S)-L5通过配体交换生成活性三价钴催化剂,在碱的作用下与底物发生碳氢键活化生成Int-1-1。此时在由配体诱导形成的手性口袋里,氧杂苯并降冰片烯2a专一取向的与Int-1-1采取外向型(exo-)配位模式生成Int-1-2,随后插入C-Co键生成七元环碳钴中间体Int-1-3。由于Co-C键特殊的亲核特性,对酰胺羰基发生分子内亲核进攻得到Int-1-4,后续C-N键断裂生成手性目标产物3aa与8-氨基喹啉配位的三价钴中间体Int-1-5。最终由醛淬灭8-氨基喹啉,实现钴催化剂的重生,实现催化循环。
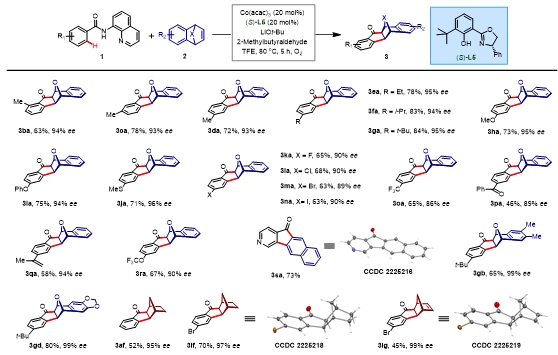
图4. 底物拓展。图片来源:J. Am. Chem. Soc.
作者对该反应的底物普适性进行探索。对各取代苯甲酰底物:在芳基上引入给电基团,反应均能以优秀产率(63-84% ee)与优秀对映体选择性(93-96% ee)得到目标手性产物,且取代基在苯环的相对位置对该反应的对映体选择性没有明显影响。但引入吸电基时,反应的对映体选择性会出现轻微地下降,以86%-90%ee得到手性产物。该反应同时能够兼容烯烃取代基和三氟甲氧基取代基。当使用异烟酰胺底物1t时,该反应会生成芳构化的产物。接着作者研究了该反应对桥双环烯烃的普适性。对于取代的氧杂苯并降冰片烯,反应的对映体选择性显著提高,能以99%ee得到手性产物,同时降冰片烯与降冰片二烯也适用于该反应。利用单晶衍射,我们确认了化合物的绝对构型,均为exo型插入产物。
鉴于对8-氨基喹啉导向的苯甲酰底物不对称碳氢键活化/[3+2]亲核环化反应的成功,作者希望能将该策略拓展至其他类型的底物。通过对苯甲酰底物详细的机理探究实验,了解到1a底物与钴催化剂发生碳氢键活化后形成的中间体(Sc)-Co-3中,底物与钴存在[阴离子(C-),阴离子(N-),中性离子(N)]的“钳型(pincer)”配位模式,并且此时配体酚羟基邻位的叔丁基诱导了桥双环烯烃分子插入的方向。由此,作者推测,当底物的导向基变为N-2-吡啶腙时,底物与钴的配位模式将变为[阴离子(C-),中性离子(N),阴离子(N-)]的pincer配位模式,并且由于“推拉效应”(push-pull effect),Salox与钴的配位模式也将改变:Salox配体中给电性较强的噁唑啉N原子处于底物中腙N原子的反位。因此,在该种配位模式下,中间体的手性口袋可以通过在噁唑啉侧链上进行修饰而调节。
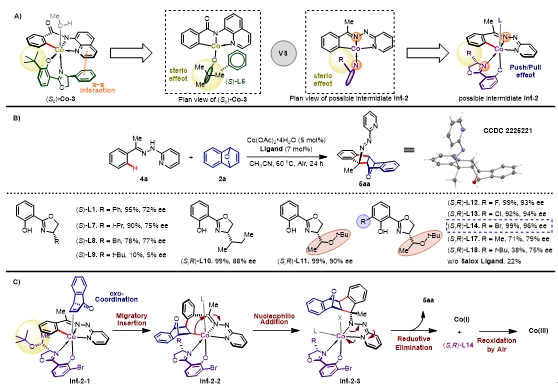
图5. 理性设计、配体筛选与机理设计。图片来源:J. Am. Chem. Soc.
为了验证设想的可行性,作者选择N-2-吡啶苯基甲基腙4a与氧杂苯并降冰片烯2a为模板底物,筛选了具有不同噁唑啉侧链的Salox配体。根据实验结果发现:当侧链叔丁基时,反应产率与对映体选择性显著下降,侧链为苯基、异丙基、苄基时反应只能以72-77% ee得到手性产物;当使用仲丁基时,反应的产率与对映体选择性都有明显提高;进一步使用(S)-1-叔丁氧基乙基[(S,R)-L11]为侧链时,反应能得到90% ee的手性产物。在(S,R)-L11的基础上对芳基进行修饰,当使用(S,R)-L14时,反应能以99%的产率,96% ee得到目标产物。
在最优条件下,作者考察该反应的底物普适性。对于芳基腙底物各类取代基均能兼容。当在对位无论引入给电基,反应均能保持高产率(85-99%)与对映体选择性(95-97% ee)。当底物具有联芳结构时,反应仍能够99% 的产率和优秀的对映体选择性95% ee得到目标产物。当在间位引入取代基甲基或氯时,会使反应产生区域选择性,碳氢键活化更倾向于发生在位阻小的碳氢键上。虽然该反应产率对芳基邻位位阻较为敏感,但能保持高对映体选择性。同时,该反应能兼容噻吩杂环。对导向基侧链进行修饰时,反应仍然能保持高对映体选择性,并且,通过改变导向基侧链,制备了含氧杂桥双环骨架的手性多元并化合物。同时,作者将底物通过酰胺键或醚键与功能分子二茂铁、药物分子吉非罗齐和天然产物香叶醇进行联接,反应仍能展现出高反应性(70-85%)与高对映体选择性(96-98% ee)。
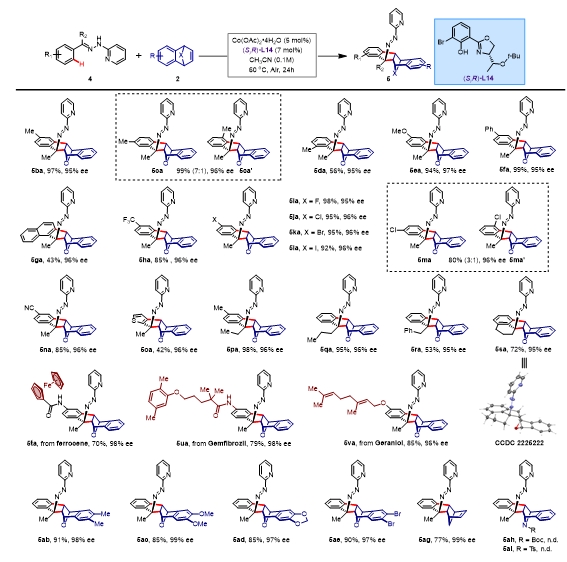
图6. 底物拓展。图片来源:J. Am. Chem. Soc.
为了进一步提升手性桥双环产物的结构多样性,作者对反应产物进行合成转化。首先,通过重结晶得到>99% ee的光学纯芳基甲酰的反应产物3aa。使用四氢铝锂可以立体专一性地将3aa还原为具有五个连续手性中心的手性醇6,且其d.r.值>20: 1,并通过单晶衍射确定了其绝对构型。接着利用缩合剂将6分别与药物分子萘普生(7)、丙磺舒(8)和材料分子DTT-2,6-dicarboxylic acid(9)缩合联接。
对于芳香腙为底物的反应,作者对其进行克级规模制备,反应规模扩大至5mmol时,反应仍能以93% 产率,95% ee得到的5aa。随后使用Raney Ni与水合肼脱除导向基,得到具有五个连续手性中心的手性胺化合物10。10可以在缩合剂的作用下与N-Boc-D-缬氨酸、薄荷醇分子通过酰胺键连接。
最后,将制备4a的原料:苯乙酮与2-吡啶肼直接投入标准反应条件中与氧杂苯并降冰片烯2a发生钴催化不对称碳氢键活化/[3+2]亲核环化反应,接着不纯化直接脱除导向基、并与药物分子布洛芬[(S)-Ibuprofen]通过酰胺键连接,通过三步反应一次柱层析,最终能以59%产率,97% de得到手性化合物13。
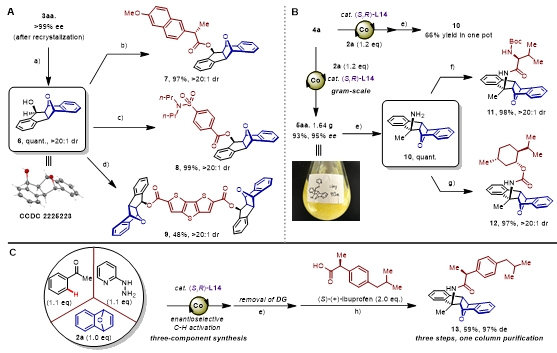
图7. 合成应用。图片来源:J. Am. Chem. Soc.
综上,浙江大学化学系史炳锋教授团队报道了Co(II)/Salox催化不对称碳氢键活化/[3+2]亲核环化反应高效、高立体专一地构建手性[2.2.1]-桥双环结构(高达99%产率,99% ee),同时一步构建连续4-5个手性中心。机理实验研究表明,配体与底物围绕三价钴中心“手性配位自组装”形成的“手性口袋(chiral pocket)”是该策略优秀的立体专一性的关键。该策略为开发具有功能性的手性桥双环化合物的制备提供新的思路与方法。
该论文的通讯作者是浙江大学史炳锋教授和百人计划研究员姚启钧,第一作者是2022级博士生黄凡芮,张鹏等同学共同参与完成该课题。
Cobalt-Catalyzed Domino Transformations via Enantioselective C–H Activation/Nucleophilic [3+2] Annulation toward Chiral Bridged Bicycles
Fan-Rui Huang, Qi-Jun Yao*, Peng Zhang, Ming-Ya Teng, Jia-Hao Chen, Lu-Chen Jiang, and Bing-Feng Shi*
J. Am. Chem. Soc.2024, DOI: 10.1021/jacs.4c04623
作者简介:
史炳锋,浙江大学求是特聘教授,博士生导师,国家杰出青年基金获得者。2001年本科毕业于南开大学化学系;2006年博士毕业于中科院上海有机所;2006-2010年先后在University of California at San Diego, The Scripps Research Institute进行博士后研究;2010年5月加入浙江大学化学系。先后获得国家自然科学基金优秀青年项目(2014)和杰青项目(2019)资助,入选浙江省万人计划(2018)、教育部青年人才计划(2017)和钱江人才(2013),曾获药明康德生命化学研究奖,日本化学会Distinguished Lectureship Award, Thieme Chemistry Journal Award,Gordon Research Conference主席奖,罗氏化学创新奖和明治乳业生命科学奖等奖励。现任美国化学会The Journal of Organic Chemistry副主编,浙江省化学会常务理事,中国化学会有机化学专业委员会、物理有机和磷化学专业委员会委员,以及《化学学报》、《有机化学》、《高等学校化学学报》、Chemical Research in Chinese Universities和Green Synthesis & Catalysis等期刊编委。主要从事惰性碳氢键的精准催化转化、不对称催化及天然产物和药物活性分子的合成研究。
史炳锋教授课题组主页:
https://person.zju.edu.cn/bfshi
文 字:史炳锋教授课题组
编 辑:黄珍珍、张维娅
审 核:林旭锋
终 审:丁立仲