
史炳锋教授课题组JACS:钴催化不对称C-H键活化插炔环化及其在生物活性分子模块化、不对称合成中的应用
C1-手性四氢异喹啉(c-THIQs)作为一类重要的生物碱骨架广泛存在于天然产物和药物活性分子中,因此,该手性结构的绿色高效合成备受关注。近年来,过渡金属催化的不对称碳氢键活化反应为复杂天然产物分子的构筑提供了一种直接、高效的合成策略。不过,现有的相关应用报道大多依赖于钯、铑等贵金属催化剂,而基于地球丰产3d过渡金属催化的策略则发展较为缓慢,根本原因在于相应手性配体体系的匮乏,实现3d过渡金属催化的不对称碳氢键活化仍存在较大的挑战。
我系史炳锋教授团队长期致力于发展绿色高效的廉价金属催化的不对称碳氢键活化新方法,自2019年以来,团队围绕廉价金属钴催化的不对称碳氢键活化,进行了系统而深入的探索。最近,史炳锋教授团队基于对金属钴配位模式的理解,提出了无Cp(Cyclopentadiene)配位的正八面体钴催化不对称碳氢键活化新策略,发展了两组新型高效的手性配体催化体系,实现了廉价金属钴催化的不对称碳氢键活化。2021年,他们率先发展了Co(II)/螺环手型磷酸(SPA)接力催化体系,实现了螺中心手性的构建(Angew. Chem. Int. Ed. 2021, 60, 23187)。随后,他们又发展了新型Co(II)/Salox(salicyloxazoline,水杨基噁唑啉)催化体系,该新型催化体系采用廉价易得的二价钴盐作为催化剂和合成简便且方便改造的Salox手性配体,在体系中原位氧化生成八面体手性Co(III)催化剂,高效地实现了不对称碳氢键活化/环化反应,并成功地分离和表征了系列正八面体手性Co(III)中间体,阐明不对称碳氢键活化的手性控制机理(Angew. Chem. Int. Ed. 2022, 61, e202202892)。随后他们将该催化模式成功应用于双轴手性化合物的构建(Angew. Chem. Int. Ed. 2022, 61, e202208912)、膦酰胺不对称碳氢键脱氢烷氧基化和胺化(Angew. Chem. Int. Ed. 2022, 61, e202210106; Angew. Chem. Int. Ed. 2023, 62, e202302964)及首例对映和区域选择性电氧化钴催化C-H/N-H环化反应(Angew. Chem. Int. Ed. 2023, 62, e202218533)、手性二芳基甲胺的构建(Angew. Chem. Int. Ed. 2023, 62, e202304706)和联芳轴手性骨架的构建(Angew. Chem. Int. Ed. 2023, 62, e202310004)。近日,该团队成功地将Co(II)/Salox催化体系拓展应用于生物活性分子的模块化、不对称合成,相关成果近期在线发表于J. Am. Chem. Soc.上。
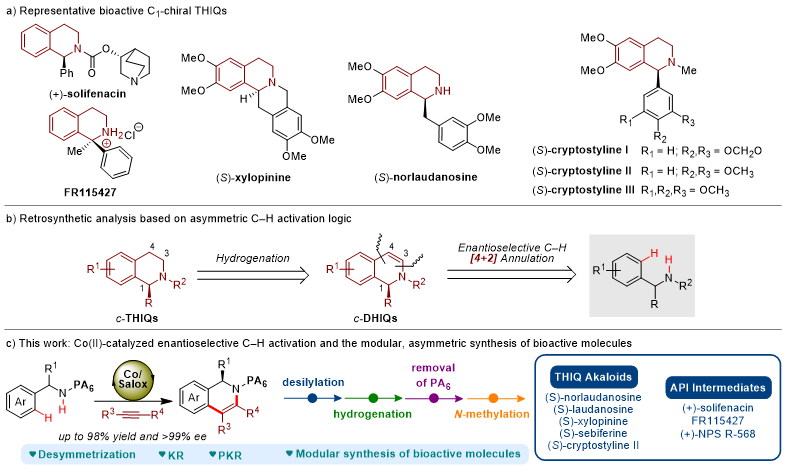
图1. Co(II)/Salox催化不对称插炔C-H/N-H环化反应。图片来源:J. Am. Chem. Soc.
受先前研究成果的启发,作者设想将Co(II)/Salox催化体系应用于C1-手性四氢异喹啉骨架的构建。基于逆合成分析,作者设想利用α-取代芳基吡啶甲酰胺与易得的炔烃进行不对称C-H键活化/插炔环化反应直接构建C1-手性二氢异喹啉骨架(c-DHIQs),再经一步氢化还原反应获得目标C1-手性四氢异喹啉骨架。然而,面临的挑战有:1)基于吡啶甲酰基(PA)导向的碳氢键活化反应往往效率较低,需要较高的反应温度和较长的反应时间;2)PA导向基团是否能与Salox手性配体兼容形成有利的手性环境;3)C1-手性二氢异喹啉类化合物在氧化条件下易芳构化,这将导致手性碳C1的消失。作者成功基于课题组发展的Co(II)/Salox催化体系,实现了不对称C-H活化/插炔环化反应,并将该策略应用于四氢异喹啉类生物碱(S)-norlaudanosine、(S)-laudanosine、(S)-xylopinine、(S)-sebiferine和(S)-cryptostyline II及药物活性分子(+)-solifenacin、FR115427和(+)-NPS R-568关键手性中间体的模块化、不对称合成(图1)。
首先,作者以α-苯基吡啶甲酰胺1a和二苯基乙炔2a作为模型底物,进行了相关反应条件筛选。筛选结果表明,最优反应条件是:Co(OAc)2•4H2O(10 mol%)作为催化剂,(S)-L1(15 mol%)作为配体,NaOPiv•H2O(50 mol%)作为添加剂,Mn(OAc)2•4H2O(20 mol%)与氧气作为共氧化剂,在MeOH(0.1 M)溶剂中90 ℃反应24小时后,可以92%的收率、95%的对映选择性得到产物3aa。
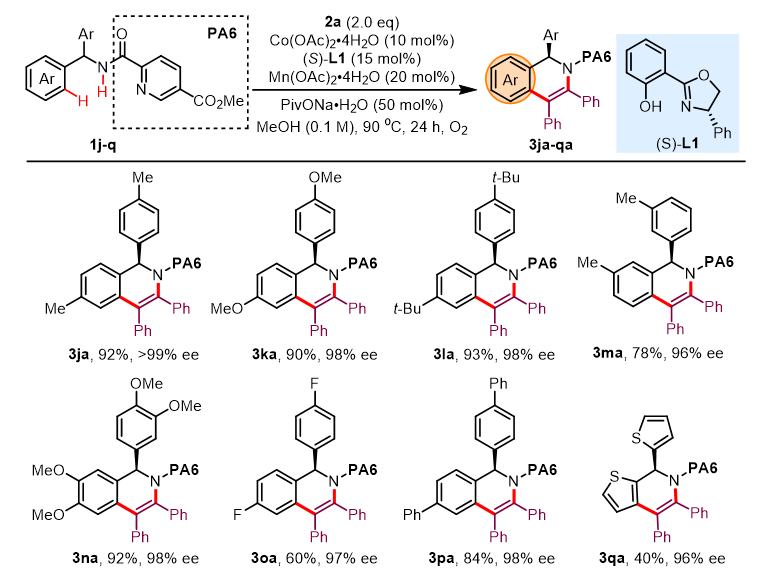
图2. α-芳基吡啶甲酰胺底物范围。图片来源:J. Am. Chem. Soc.
在获得上述最佳反应条件后,作者首先对α-芳基吡啶甲酰胺底物2的范围进行了考察(图2)。各类含供电子基、吸电子基的α-芳基吡啶甲酰胺底物均能有效兼容(3ja-3pa, 60-93%, 96->99% ee),同时α-噻吩杂环的吡啶甲酰胺底物也能顺利兼容,以中等的收率得到产物3qa(40%, 96% ee)。随后,作者通过X射线衍射分析确定了3qa的绝对构型,其他产物的立体构型可类比得知。
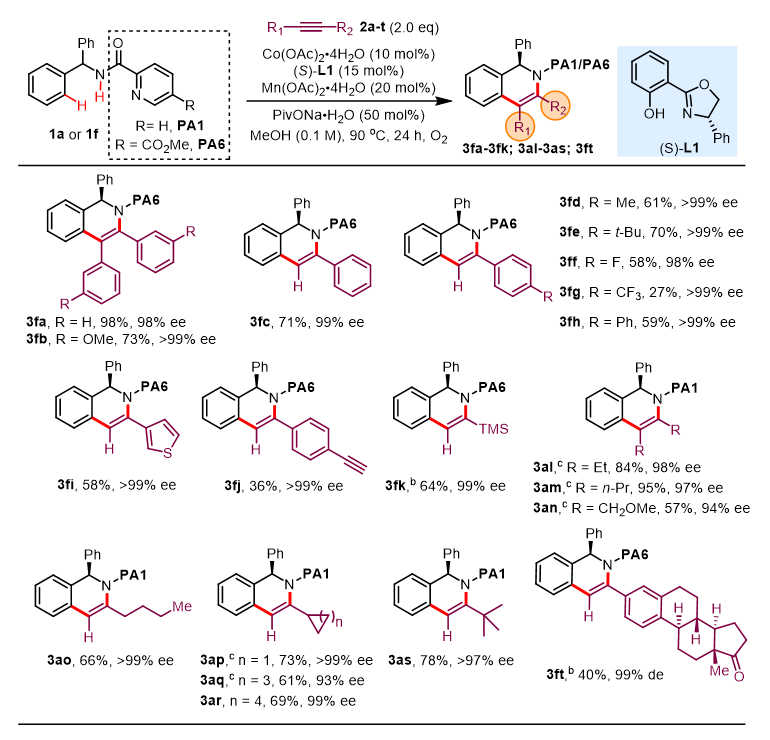
图3. 炔烃底物范围。图片来源:J. Am. Chem. Soc.
作者接着对炔烃的普适性进行了考察(图3)。该反应体系对于含供电子基、吸电子基的芳基乙炔和3-乙炔基噻吩都可以给出中等到优秀的产率以及优秀的对映体选择性(3fc-3fi, 98->99% ee)。对于大位阻的乙炔基三甲基硅烷(2k),在提高反应温度后也能顺利兼容。同时,反应体系也能较好地兼容对称型烷基类炔烃(3al-3an)。值得一提的是,Co/Salox催化体系不仅能给出优异的对映选择性,同时还具备优异的区域选择性控制,当使用端炔时,能获得高立体选择性的专一异构体产物(3fc-3fk、3ft、3ao-3as,93->99% ee)。
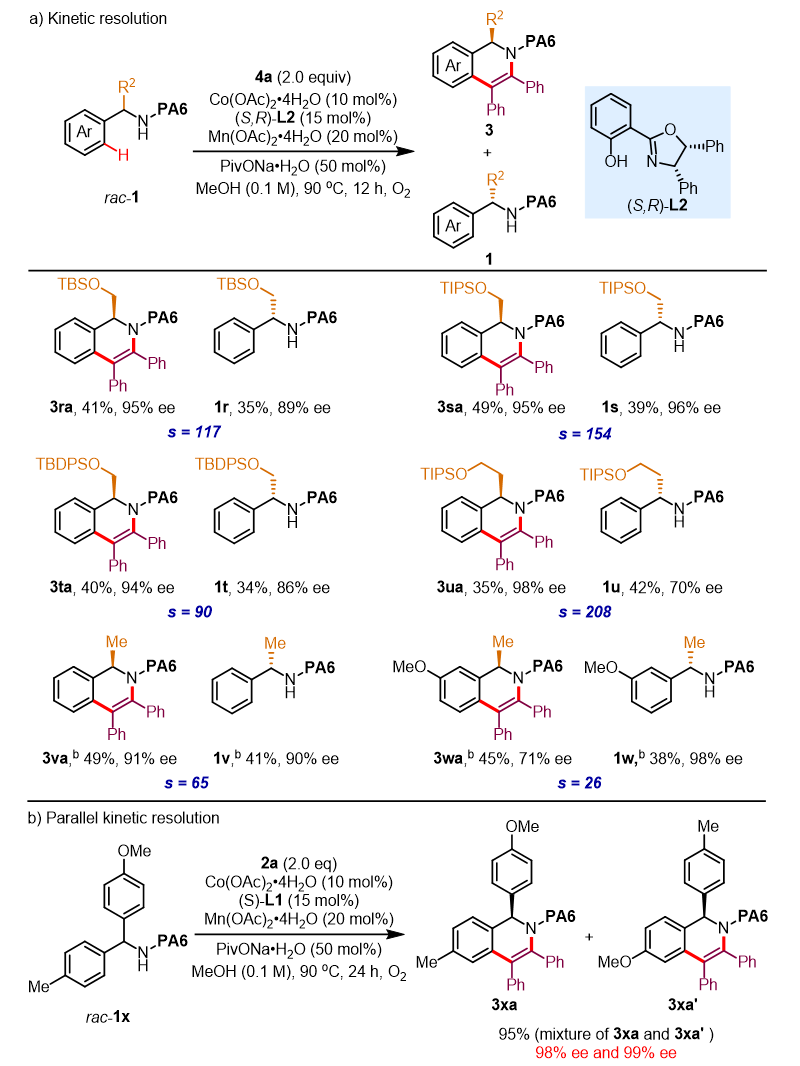
图4. 动力学拆分与平行动力学拆分底物范围。图片来源:J. Am. Chem. Soc.
令人惊喜的是,该Co(II)/Salox反应体系还能实现外消旋α-取代苄酰胺底物的动力学拆分(图4a)。在微调反应条件后,作者使用(S,R)-L2作为手性配体,成功地对一系列外消旋羟基保护的氨基醇衍生物进行了动力学拆分,获得了中等至优异对映选择性的环化产物3和手性氨基醇衍生物1(3ra-ua, 94-98% ee; 1r-u, 70-96% ee)。此外,反应体系也可实现平行动力学拆分过程(图4b)。
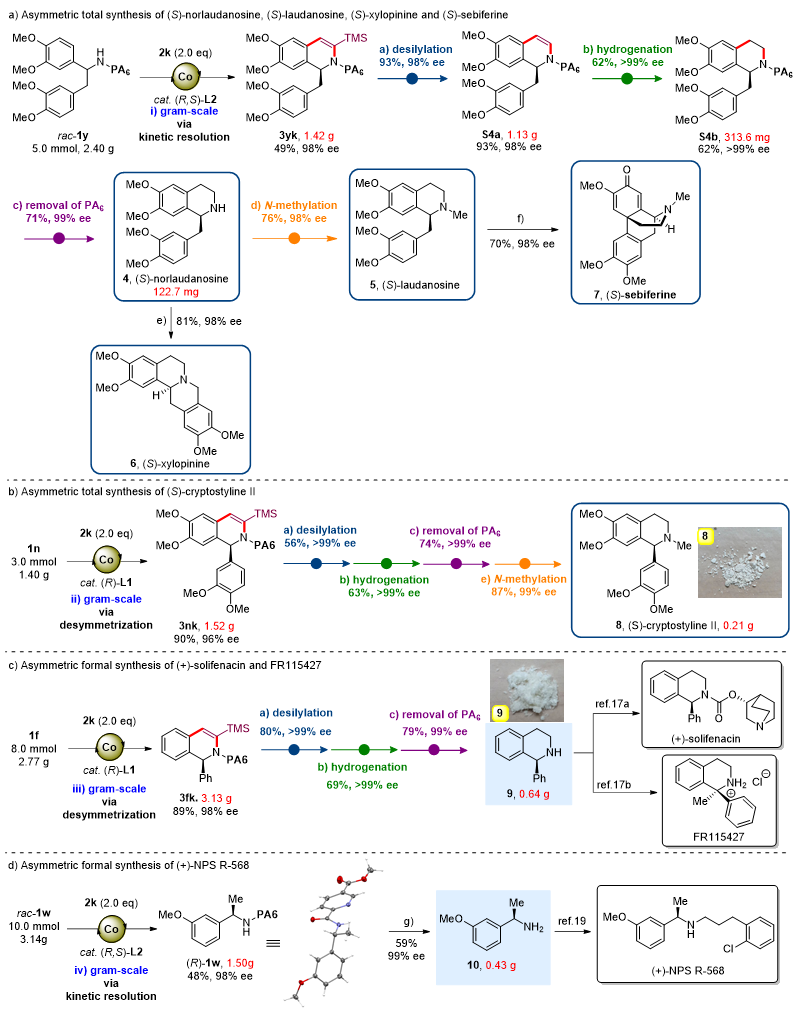
图5. 合成应用。图片来源:J. Am. Chem. Soc.
最后,为了展示Co(II)/Salox催化体系的合成应用潜力,作者利用Co(II)/Salox催化的不对称C-H键活化/插炔环化作为关键手性合成步,通过去对称化和动力学拆分过程,高效、高对映选择性制备了系列C1-手性二氢异喹啉衍生物。后续经由硅基脱除、氢化还原、导向基团移除和甲基化等步骤,实现了含C1-手性四氢异喹啉骨架天然产物(S)-norlaudanosine、(S)-laudanosine、(S)-xylopinine、(S)-sebiferine和(S)-cryptostyline II及活性分子(+)-solifenacin, FR115427和(+)-NPS R-568关键手性中间体的模块化、高效不对称合成。
综上,史炳锋教授团队报道了Co(II)/Salox催化的不对称C-H键活化/插炔环化反应。该反应体系以廉价易得的钴盐作为催化剂、易制备的水杨基噁唑啉作为手性配体,条件温和、立体选择性高,为C1-手性二氢异喹啉骨架的高效绿色合成提供了新的方法。
该论文的通讯作者是史炳锋教授和姚启钧博士,第一作者是吴勇杰博士,2019级直博生陈嘉豪,2020级直博生滕茗芽等同学共同参与完成该课题。
文 字:史炳锋教授课题组
编 辑:张维娅
审 核:黄珍珍、林旭锋
终 审:丁立仲