
曹亮研究员课题组JACS:基于机器学习构建合金纳米催化剂“组分-尺寸”催化活性“等高图”
曹亮研究员与合作者美国约翰霍普金斯大学Tim Mueller教授合作在基于机器学习方法的多相催化剂理性设计方面取得进展。研究成果以“合金纳米颗粒的催化活性图(Catalytic Activity Maps for Alloy Nanoparticles)”为题,于2023年3月27日在线发表于JACS。论文链接:https://doi.org/10.1021/jacs.2c13607 。
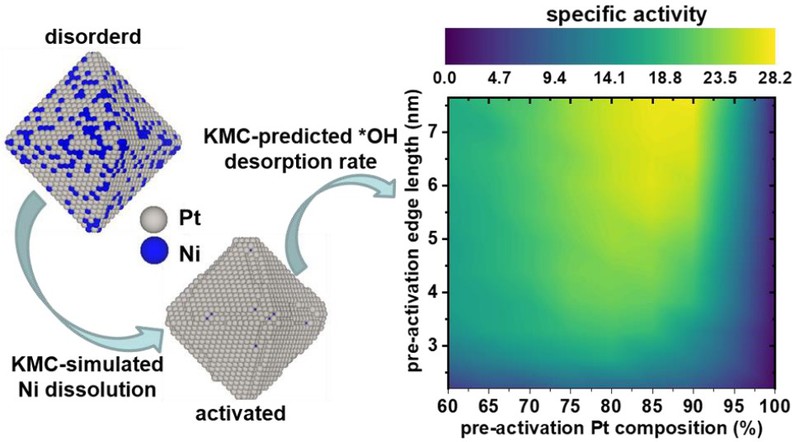
图1.固溶相无序Pt-Ni纳米颗粒的“组分-尺寸”催化活性“等高图”。
合金纳米颗粒因其高表面积比、结构和性能可调等性质,非常适合作为催化剂。尽管各种实验合成策略(如核壳、掺杂等)已被用于提高合金纳米催化剂的催化活性和稳定性,但由于实验的昂贵成本和耗时较大的限制,通过理论计算对其进行理性设计具有极大的发展前景。然而,密度泛函理论(DFT)所需计算资源将随着体系价电子数量以N3的方式增加,这极大地限制了DFT可实际模拟的纳米颗粒直径(2-3nm),远低于实验合成中的典型直径(4-10 nm)。此外,当通过热力学或动力学来预测合金纳米颗粒的结构及其催化性能时,需要得到相空间中大量可能原子排列结构的能量(不同构型之间的能量差异可以在meV/atom的量级),DFT方法对此同样无能为力。
针对合金纳米催化剂理性设计的难题,研究人员通过DFT计算和机器学习方法的有机结合,发展了一个能够同时预测合金纳米颗粒的结构、原子排列、中间产物吸附能、“吸附物-吸附物”相互作用的替代模型(surrogate model),从而研究合金纳米颗粒及其表面吸附物在催化反应发生条件下的动态演化过程。基于此,本文发展了一个能够快速、精确预测实验相关尺寸(5-10 nm)合金纳米颗粒结构及其催化活性的新方法,进而在“合金组分-颗粒尺寸”构成的二维网格上成功构建了催化活性的“等高图”。 本文以用于燃料电池阴极氧还原反应(ORR)的Pt-Ni纳米颗粒催化剂为研究对象,预测得到的催化活性“等高图”表明ORR面积活性在组成为Pt0.85Ni0.15、尺寸大于5.5 nm时具有最大值(图1),充分展示了该方法的可行性。
这项工作中发展的新方法不仅在Pt-Ni合金作为ORR催化剂上得到了展示,也有望在其他合金体系(或多组分材料)和多相催化反应中得到广泛的应用。相信该方法能为合金纳米催化剂的理性设计提供一套有效的工具。
本文第一作者为化学系曹亮研究员,曹亮研究员和约翰霍普金斯大学Tim Mueller教授为共同通讯作者。本课题受国家自然科学基金(22203072)和中央高校基本科研业务费(226-202200167)的资助。
曹亮研究员个人主页:https://person.zju.edu.cn/caoliang