
化学系丁寒锋课题组:木藜芦烷二萜(‒)-Rhodomollanol A的首次不对称全合成
木藜芦烷二萜广泛分布于杜鹃花科植物中,迄今为止已有超过150个家族成员被分离鉴定出来。除了高细胞毒性,这类分子还体现出多种多样的生物活性,如调节钠离子通道、止痛、镇静、杀虫和拒食活性等。从结构上看,木藜芦烷二萜共同拥有一个[5,7,6,5]四环母核,其中包含与对映-贝壳杉烷二萜相似的双环[3.2.1]辛烷骨架,分子内含有7−12个手性中心,并体现出较高的氧化态(图1a)。由于极具挑战性的复杂骨架结构和潜在的重要生物活性,木藜芦烷二萜引起了合成化学家和药物化学家的广泛关注(Phytochem. Rev., 2013, 12, 305)。然而,到目前为止仅有两例关于其家族成员的全合成报道(Grayanotoxin II:Tetrahedron Lett., 1972, 13, 3087 & Tetrahedron Lett., 1976, 17, 553;Grayanotoxin III:J. Org. Chem., 1994, 59, 5532),且均步骤较长(>38步),效率低下(<0.05%总收率)。近期,耶鲁大学的Newhouse课题组也完成了C1位具有β-H的家族成员Principinol D的全合成(J. Am. Chem. Soc., 2019,141, 8088)。由于此类结构在木藜芦烷家族中极为稀少,该合成策略在应用上存在一定的局限性,新颖、高效的策略亟待发展。
经典的木藜芦烷二萜不同,(‒)-Rhodomollanol A具有更高合成挑战性的[3,5,7,5,5,5]六环体系,其中包含一个独特的7-氧杂双环[4.2.1]壬烷核心骨架(蓝色显示)和11个连续的手性中心(图1a)。该天然产物的发现进一步丰富了木藜芦烷二萜的骨架类型。(‒)-Rhodomollanol A与经典木藜芦烷家族成员(如Rhodojaponin III和Rhodomollein XXXI等)在生源上密切相关(Org. Lett., 2017, 19, 3935),后者则被认为可能是由对映-贝壳杉烷经AB环系的重排转化得到(Agric. Biol. Chem., 1981,45, 1281)。因此,开展(‒)-Rhodomollanol A的全合成研究对于增进对该分子骨架生源合成途径的理解具有十分重要的意义。
丁寒锋课题组于2016年启动了针对木藜芦烷二萜的系统性合成研究课题。经过几年的持续努力,他们于2019年发展了一类Ti(III)介导的环氧还原开环/Beckwith–Dowd重排串联反应,在复杂体系中实现了从双环[2.2.2]辛烷骨架向双环[3.2.1]辛烷骨架的高效转化,并以此为关键步骤高效完成了木藜芦烷二萜Rhodomolleins XX和XXII的首次全合成(Angew. Chem. Int. Ed., 2019, 58, 8556)。近期,受生源合成途径的启发,该课题组巧妙地借助三个串联反应接力的方式实现了(‒)-Rhodomollanol A的首次不对称全合成。
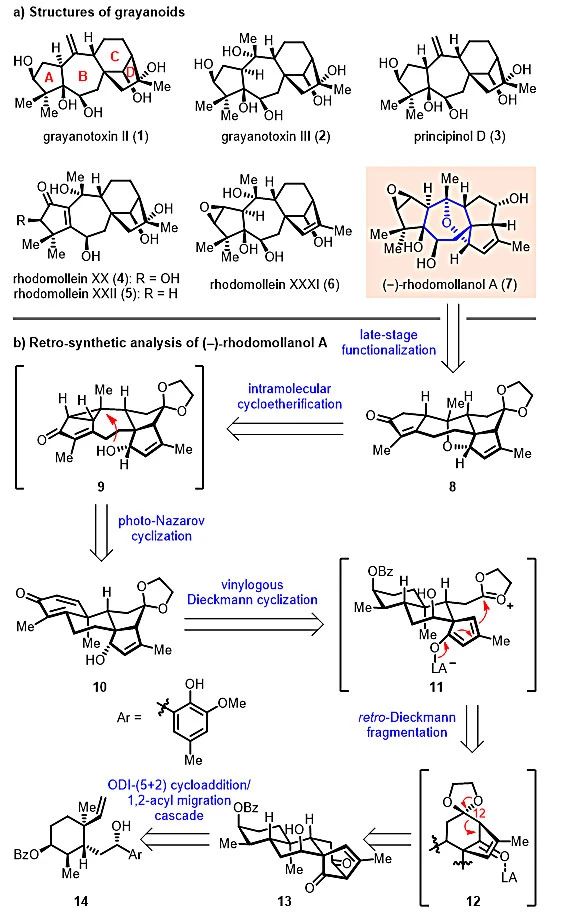
图1. 木藜芦烷家族代表性分子的结构及(‒)-Rhodomollanol A的逆合成分析
逆合成分析如图1b所示:他们考虑(‒)-Rhodomollanol A可由中间体8经后期官能团转化得到,而8结构中的7-氧杂双环[4.2.1]壬烷骨架则可从环己二烯酮中间体10出发,通过紫外光促进的山道年重排/分子内环醚化串联反应来构建。关于10的合成,研究人员计划运用一种新颖的骨架重排反应,在Lewis酸促进下,将中间体13的C12位酮羰基通过缩酮保护得到激活,从而可引发后续的逆-Dieckmann裂解/插烯-Dieckmann环化串联反应,构建出双环[3.3.0]辛烷骨架。最后,含有双环[3.2.1]辛烷骨架的13可由烯基苯酚中间体14经该课题组前期所发展的ODI-[5+2]环加成/频哪醇重排串联反应得到。
2017年,丁寒锋课题组发展了一类全新的氧化去芳香化促进(ODI-)的[5+2]环加成/频哪醇重排串联反应(图2a)。从简单易得的烯基苯酚出发,在有机三价碘试剂(如PIFA)氧化下,以六氟异丙醇为溶剂,可一步生成3个环和3个立体中心(其中1个为季碳),以中等至良好收率快速构建出双环[3.2.1]辛烷骨架结构。作为该方法学的初步应用,该课题组从已知的手性萘烷衍生物出发,以该串联反应作为关键步骤,并结合Eschenmoser−Tanabe裂解、逆-aldol/aldol串联反应和Schenck ene反应,以14−16步、6.0%−6.8%的总收率实现了Pharicin A、Pharicinin B、Pseurata C和7-O-Acetylpseurata C等4个高氧化态对映-贝壳杉烷二萜的首次不对称全合成(图2b,J. Am. Chem. Soc., 2017, 139, 6098)。值得注意的是,研究人员通过调整前体(烯基苯酚)结构中C7位羟基的立体构型有效控制了ODI-[5+2]环加成/频哪醇重排串联反应的优势过渡态,实现了其高立体选择性。最近,他们又基于该[5+2]环化串联反应完成了另一类四环二萜滨海孪生花素Stemarin的不对称全合成(Org. Lett., 2020, 22, 1426)。
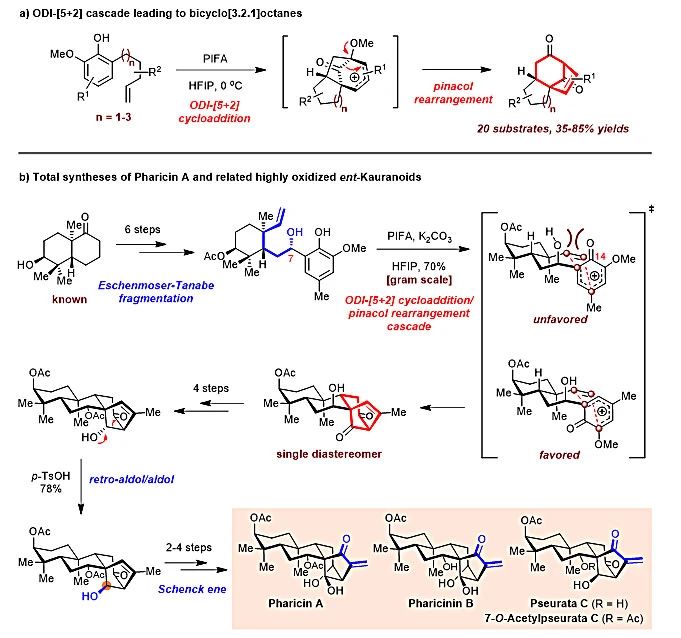
图2. ODI-[5+2]环化串联反应及高氧化态对映-贝壳杉烷二萜的不对称全合成
在针对(‒)-Rhodomollanol A的合成过程中,首先是关于环己二烯酮中间体27的合成(图3):从商业可得的萘烷衍生物16(95% ee)出发,经7步常规转化得到烯醛15。后者与由17转化得到的有机芳基锂试剂发生1,2-加成,以78%的收率、2.7:1的dr值得到烯基苯酚18及其C7-异构体的混合物。随后,18经THP保护和MOM脱除后,在三价碘氧化下发生预想的ODI-[5+2]环加成/频哪醇重排串联反应,转化为13。研究人员认为该反应的立体选择性由C7-羟基的立体构型控制。受C7-OTHP与C14-酮羰基之间的1,3-斥力影响,反应主要经过渡态19进行。必须指出的是,在该反应中THP保护基的采用是经过大量筛选后的最优选择。此外,如C7-羟基不经保护,反应将经历活泼的邻亚甲基醌中间体,从而诱发一系列环化,转化为副产物21。获得13后,在TMSOTf、(TMSOCH2)2条件下,顺利发生逆-Dieckmann裂解/插烯-Dieckmann环化串联反应,在克级规模高效得到22,再经Barton脱氧反应转化为23。随后,研究人员在对23的C14-酮羰基进行还原尝试时遇到了极大的困难,超过50余种不同的还原条件均无法获得α构型的醇羟基。这些结果表明,可能是由于C20-角甲基的存在,使得C14-酮羰基的β-面具有较大的空间位阻。因此,研究人员希望通过改变D环的结构来对构象进行微调。围绕这一思路,首先将还原产物24进行诱导环氧化,造出β-环氧。接着,通过进一步氧化,转化为环己二烯酮中间体26。此时,再进行1,2-还原,即可以6:1的dr值成功得到重排前体27。
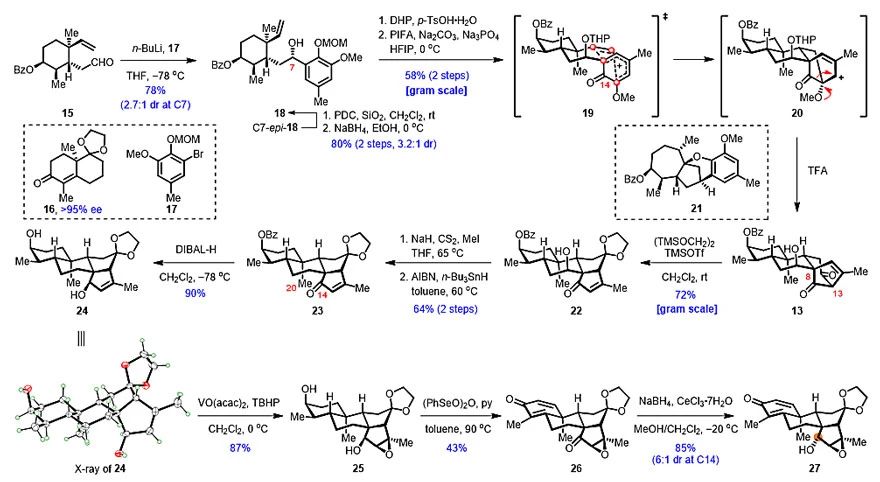
图3. 环己二烯酮中间体27的合成
接下来,研究人员对紫外光促进的山道年重排/分子内环醚化串联反应进行了初步尝试(图4)。令人失望的是,反应以82%的收率专一性转化为螺环副产物30。该结果表明,反应在经历中间体29A时快速发生了C14-羟基对环丙烷的开环(path a)。受Broka课题组工作的启发,研究人员合成了C2位带有甲酯的环己二烯酮中间体28。在与之前相同的条件下,该反应可以70%的收率得到预期产物32,再经脱羧可最终转化为33。然而,总体而言,这一策略显得比较迂回,而且总收率仅为14%,显然很难将料往前推进。因此,需要寻找其它更为高效的方法。研究人员设想,能否将重排前体结构中的C14-羟基首先用一个合适的保护基保护起来。接着,在反应过程中,利用这个保护基对醋酸的不稳定性,做到缓慢释放。在形成中间体29A’时,由于保护基的存在,可以抑制C14位羟基对环丙烷的开环,使得29A’有机会转化到环丙烷中间体29B’(path b),此时再脱除保护,就能一步得到所需产物33。围绕这一想法,他们考察了多种保护基。最后发现,当使用TMS时,反应可预期发生,以56%的收率主要得到目标产物。同时,反应中也监测到了少量的30、34和35。这些副产物的生成足以说明这一反应存在的巨大挑战性和TMS预先保护C14-羟基的重要性。

图4. 光促山道年重排/分子内醚化串联反应研究
得到33之后,研究人员开始对(‒)-Rhodomollanol A进行后期合成(图5)。他们发现,在NaH和DMSO所形成的碱(“dmsyl” anion)存在下,以干燥的HMPA作为溶剂,可以区域选择性在33的C6位发生去质子,再经C4-烷基化,得到目标产物37。值得注意的是,这一转化虽然在[6,6]骨架上得到了较为充分的研究,以往文献对于[5,7]环系需要首先在C2位引入占位基团才能得以迂回实现,而这将不可避免的降低整体的转化效率。接着,对C5-C6双键进行双羟化,并顺势进行丙酮叉保护,得到38。接下来需要翻转C1位的立体化学,为此首先将38转化为烯酮39。接着,研究人员尝试了大量的1,4-还原条件,发现均无法解决这一问题。最终他们发现,将39转化为腙,再经立体选择性还原,得益于C19位假直立键甲基的阻挡作用,可以顺利获得α-构型的烯丙基二氮烯中间体40。然后,在严格除氧的溶剂中加热进行“alkene walk”,以45%的收率得到产物41。通过Ti(III)还原的方式打开环氧,再经Chugaev消除转化回双键,并脱除保护基,以3.3:1的比例获得42和43。43可在酸性条件下发生环醚化,重新转化为42。最后,利用C5-羟基对C2-C3双键立体选择性的诱导环氧化,再经立体选择性还原C12位酮羰基,顺利实现了(‒)-Rhodomollanol A的全合成。
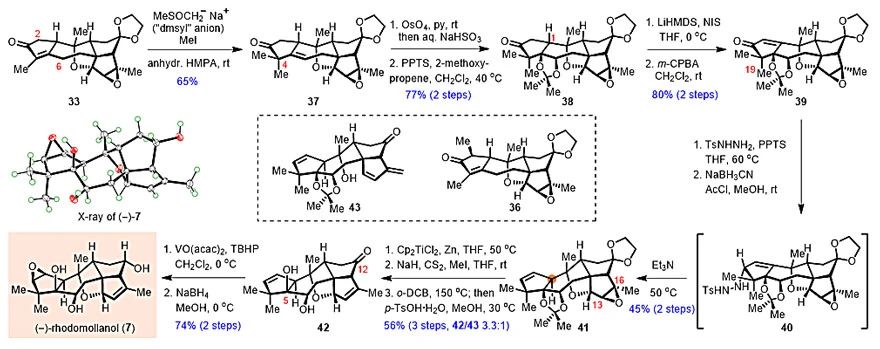
图5. (‒)-Rhodomollanol A的后期合成
小结
该项工作从易得原料烯醛和芳基溴出发,完成了具有重排骨架的复杂木藜芦烷二萜(‒)-Rhodomollanol A的首次不对称全合成研究。其中的关键步骤包括:ODI-[5+2]环加成/频哪醇重排串联反应、逆-Dieckmann裂解/插烯-Dieckmann环化串联反应和“缓释保护基”参与的光促山道年重排/分子内环醚化串联反应。这一工作的开展进一步验证了ODI-[5+2]环加成/频哪醇重排串联反应的巨大应用潜力,同时也为探究(‒)-Rhodomollanol A可能的生源合成途径提供了关键证据。相关成果发表于知名期刊J. Am. Chem. Soc.上,通讯作者为丁寒锋教授,博士生高建红和饶培榕为共同第一作者。
原文:Total Synthesis of (−)-Rhodomollanol AJianhong Gao, Peirong Rao, Kaixiang Xu, Shuaifeng Wang, Yufei Wu, Chi He, Hanfeng Ding* J. Am. Chem. Soc., 2020, 142, 4592-4597, DOI: 10.1021/jacs.0c00308
丁寒锋教授课题组主页链接https://person.zju.edu.cn/ding