近期,我系2010届校友、加拿大不列颠哥伦比亚大学助理教授还涛在Nature Methods期刊上发表研究成果:基于串联质谱的高精度分子式预测。
以下为内容转载自:“X-MOL资讯”微信公众号。
自然界中已检测到或已在实验室中合成的化合物数量达到了数百万种,尽管如此,我们对生物和环境系统中化学物质的现有认识仍然远远不够。特别是在利用组学和质谱学进行大规模化学分析的时代,在与质谱数据库匹配之后,仍存在相当一部分化学信号未被鉴定(称为化学“暗物质”)。分子式确定是阐明化学暗物质并推进其后续鉴定的第一步。然而,现有的生物信息学工具在利用质谱数据确定分子式方面仍有很多不足,表现受到质谱分辨率的限制,而且对于大分子(> 500 Da),注释性能不尽如人意,并且计算速度受到限制。
自下而上处理(Bottom-up processing)是指将单个基础组件分别指定并组装成顶层系统的过程。自下而上的方法已广泛应用于其他组学领域,如鸟枪法蛋白质组学,确定的多肽序列可以用来推测蛋白质性质。近日,加拿大不列颠哥伦比亚大学还涛教授团队提出自下而上的方法来研究串联质谱(MS/MS)信号并从中精准预测小分子化合物的分子式。该方法核心设计是注释观察到的碎片离子及其相应的中性丢失的分子式,并将它们组装成一个高层系统来确定前体分子式。图1比较了自上而下和自下而上方法的分子式预测工作流程上的区别。相比之下,本文自下而上方法具有三个明显的特点,包括:(1)产生一个可通过MS/MS解释的候选空间,(2)机器学习辅助的候选项排名,(3)假阳性发现率预估。基于此方法,作者开发了分子式预测软件“BUDDY”。
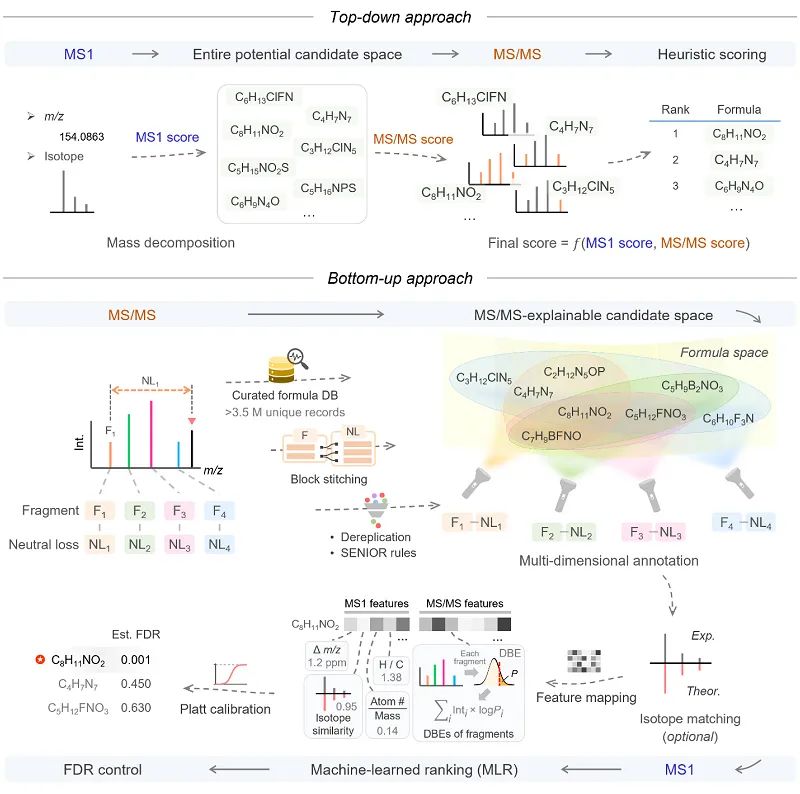
图1. 自上而下和自下而上方法的分子式预测的示意工作流程
自下而上MS/MS解析始于将查询MS/MS分解成多个碎片离子-中性损失对。每个碎片和中性损失的质量分别针对精心设计的分子式数据库(包含来自26个化学数据库超过350万个独特分子式)进行搜索,以生成分子亚式候选集。在搜索过程中,通过针对经过氢调整的分子式数据库(例如C6H7)或原始的中性分子式数据库(例如C6H6)对偶电子和奇电子(自由基)离子种类都予以考虑。接下来,每个碎片-中性损失对的分子亚式候选项被拼接在一起,以产生唯一的分子式候选空间,在其中所有分子式候选项都可以在有效的亚式级别上解释这个碎片离子-中性损失对。因此,每个碎片-中性损失对都会作为一个新的维度添加到整个MS/MS可解释候选空间的大小中。所有碎片-中性损失对贡献的候选空间被合并,去重,然后经过SENIOR规则过滤掉化学上不合理的分子式(例如CH5)。上述过程在一个汇总空间中创建了一个空间,在其中任何候选项都可以解释查询MS/MS中的至少一个峰。可选地进行MS1同位素模式匹配以生成MS1同位素相似性。以下MLR评估了候选分子式的内在属性及其在MS/MS解释中的表现,为每个候选项分配了一个MLR预测分数。然后,使用Platt校准将MLR分数转换为估计的后验概率,并相应地计算FDR估计
在实践中,通过优先考虑MS/MS可解释的候选分子式,在87.8%的总查询中可以获得较窄的搜索空间(图2)。与自上而下的方法相比,自下而上的方法将候选空间缩小了4.2%至>99.9%,平均为42.8%(中位数为40.8%)。为了进一步探究前体质量领域内的候选空间收缩,作者绘制了两个空间内所有查询的候选计数,如图2b所示。总体而言,随着前体质量的增加,两个候选空间都在扩展,但是MS/MS可解释的候选分子式空间增长速度远远低于整个空间。根据LOWESS的估计,MS/MS可解释的候选分子式空间在m/z 400处比整个空间窄7.9倍,在m/z 600处比整个空间窄12.2倍,在m/z 800处比整个空间窄20.7倍。在更高的m/z值下,两个空间之间的大小差异更加明显。即使在m/z<200的情况下,空间大小之间也存在统计学显著性(P < 2.2×10−16)。
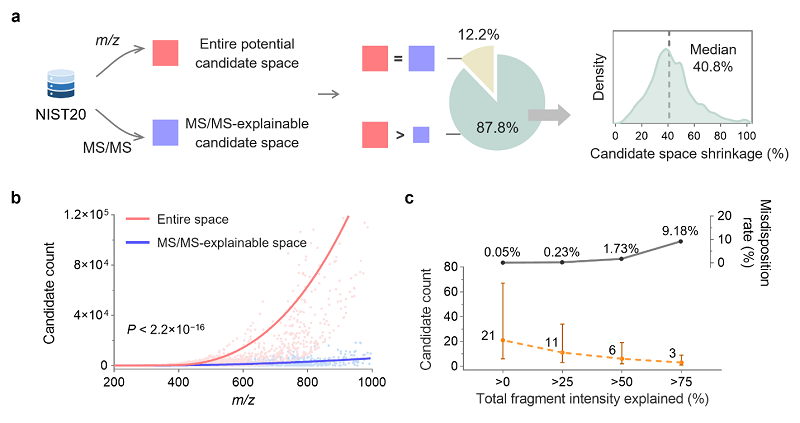
图2. 自上而下和自下而上方法的分子式预测的搜索空间比较
在包含11,667种独特分子的四个参考MS/MS库中进行评估时,与分子式确定基准工具SIRIUS相比,BUDDY的最高概率分子式(完全匹配期望的分子式)的准确性平均提高了30.1%。在使用不同MS平台收集的公共实验代谢组学数据集中,BUDDY也表现出了类似的性能结果。为了展示在大规模上发现未报告的分子式的能力,作者将BUDDY应用于155,321个反复未识别的质谱图,并在估计的FDR < 5%的情况下使得 >5,000 个不存在于化学数据库中的分子式得到可信注释。通过基于MS/MS匹配的结构类似物搜索进行的正交验证进一步确认了对未标识的分子式注释中的97.8%。最后,作为BUDDY中实验特定全局峰注释的演示,作者处理了一个比较人类粪便数据集(杂食与素食主义者),并系统注释了37个脂肪酸酰胺分子,其中有9个分子式在代谢组数据库中不存在。这些结果表明了BUDDY在探索化学“暗物质”方面的巨大优势和潜力(图3)。
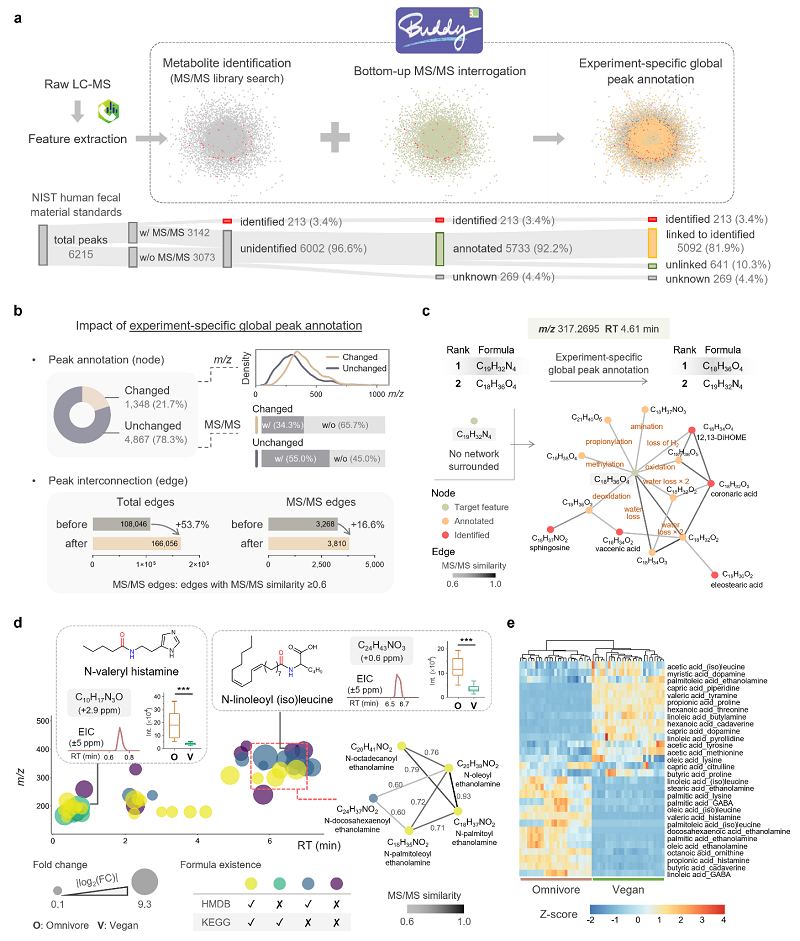
图3. BUDDY在分子式预测上的表现
最后需说明的是,BUDDY是使用C#语言编写的通用Windows平台(UWP)应用程序(图4)。它目前可在Windows操作系统(Windows 10或更高版本)上运行。该独立软件可从GitHub [1] 和Zenodo [2] 免费下载,源代码也可在GitHub [3] 上获得。同时,还涛实验室正在开发Python API版本,可以让用户将BUDDY内嵌到他们自己的软件开发中。该程序估计在今年5月份可以上线。
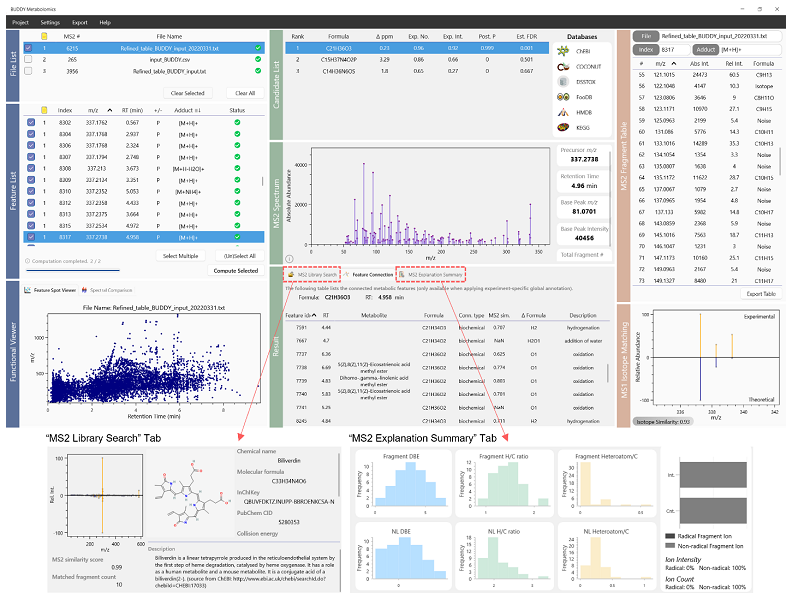
图4. BUDDY 用户界面
小结
本文提出了一种自下而上的基于MS/MS的分子式预测方法并开发了相应软件BUDDY。在自下而上的方法中,MS/MS可解释的候选空间可以比整个候选空间大大缩小(例如,在m/z 800处缩小了约20倍)。这种设计将大质量特征的计算成本从每个查询的小时级别降低到秒级。更重要的是,与自上而下的方法相比,结合MLR和FDR估计显著提高了自下而上质谱分析的性能(最高可提高68.7%的准确性),不仅能够发现已知和未知的分子式,而且可以提供注释置信度的估计。BUDDY的开发离不开整个代谢组学社区的努力,使用了高质量的MS/MS参考库、公共代谢组学数据集和开源算法。特别是,作者使用了NIST20、GNPS、MassBank和Fiehn HILIC等质谱数据库,用于训练或基准测试。这些数据库是促进软件开发的优秀资源。此外,随着更多的化学信息学工具的开发,期待对化学暗物质中未报告小分子及其生物功能提供更多的结构洞察力。
加拿大不列颠哥伦比亚大学的博士生邢世佩为第一作者,还涛教授为通讯作者。