
The asymmetric ring-opening reaction of non-strained cyclic ethers has long been a challenging problem in synthetic chemistry due to their lack of thermodynamic driving force for ring-opening. At present, the asymmetric ring-opening of non-strained saturated cyclic ethers is mainly achieved through two strategies: chiral induction and kinetic resolution, both of which have certain limitations. The chiral induction strategy requires the pre-synthesis of chiral cyclic ether precursors, involving tedious steps; the kinetic resolution strategy is limited by a theoretical maximum yield of 50%. In contrast, the enantioconvergent transformation strategy has significant advantages: it can directly start from racemic raw materials and theoretically achieve a 100% yield. Therefore, the development of an enantioconvergent ring-opening reaction for non-strained saturated cyclic ethers holds important research significance.
However, this reaction faces multiple challenges: 1. Low reactivity: non-strained cyclic ethers lack the thermodynamic driving force for ring-opening; 2. Difficult chiral control: the catalyst needs to simultaneously recognize the two enantiomeric raw materials and efficiently convert them convergently into a single chiral product.
To address the above challenges, the research team proposed a novel tandem transformation strategy that realizes ring-opening via β-O elimination mediated by cobalt-hydride species, followed by stereoconvergent hydroboration. Through this strategy, the research team achieved the first enantioconvergent ring-opening reaction of non-strained saturated cyclic ethers.
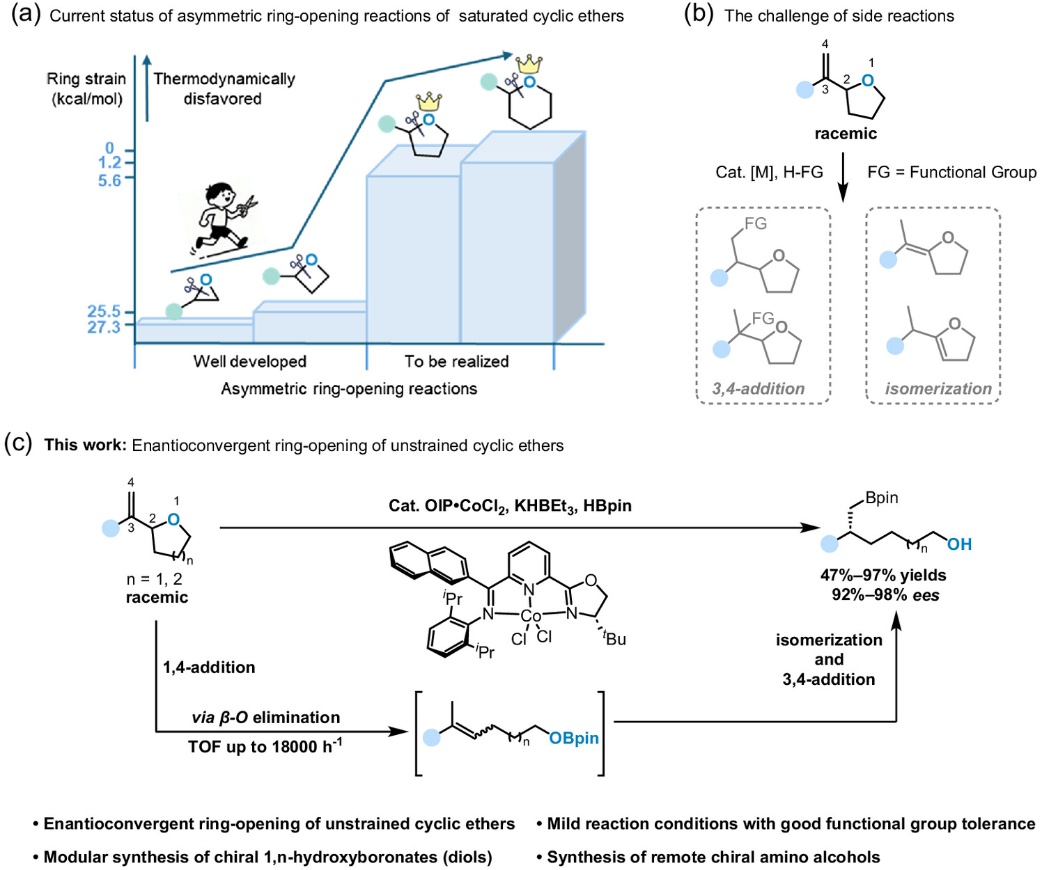
Figure 1. Background Introduction and the Work in This Paper
This method features mild reaction conditions and excellent functional group tolerance, being compatible with a variety of electron-donating/electron-withdrawing substituents, as well as methylthio, dimethylamino, silyl, ester groups and other functional groups. It also exhibits good compatibility with various fused rings, heterocycles and natural product fragments. In addition, the reaction is not only applicable to conventional non-strained (five-membered, six-membered) cyclic ethers, but also shows good compatibility with highly strained (three-membered, four-membered) cyclic ethers and moderately strained (seven-membered) cyclic ethers. The research team further realized the selective functionalization of the distal hydroxyl group of the product diols through the Mitsunobu reaction, efficiently synthesizing a series of long-chain 1,n-chiral amino alcohols, which demonstrates the practical value of this reaction. Relevant mechanistic experiments indicated that the reaction proceeds through a process where β-O elimination first causes ring-opening to form an alkene, followed by isomerization and asymmetric hydroboration of the alkene.
Based on the persistent OIP ligands developed by the research group, this study for the first time achieved the enantioconvergent ring-opening reaction of non-strained saturated cyclic ethers. This reaction not only breaks through the bottlenecks of traditional strategies such as tedious raw material synthesis and limited theoretical yield, but also provides new ideas and feasible approaches for the enantioconvergent ring-opening transformation of various heterocyclic compounds.
This achievement was published in CCS Chemistry. Kangyu Xiong, a 2023-level postgraduate student in the Department of Chemistry, Zhejiang University, is the first author of this paper, and Professor Zhan Lu is the corresponding author. This research project was supported by the National Key R&D Program of China (2021YFA1500200), the National Natural Science Foundation of China (22525109), the Natural Science Foundation of Zhejiang Province (LDQ24B020001), the Fundamental Research Funds for the Central Universities, the Forward-Looking Chemical Technology Center, the State Key Laboratory of Soil Pollution Control and Remediation Technologies, the Xinjiang Key Laboratory of Organosilicon Functional Materials, as well as the State Key Laboratory of Coordination Chemistry at Nanjing University and other projects and platforms.
CCS Chem. 2026, DOI: 10.31635/ccschem.026.202507003